Storage disease on Autopsy
| HOSP # | WARD | Histopathology | |
| CONSULTANT | Dr. Jody Rusch | DOB/AGE | 1 week Female neonate |
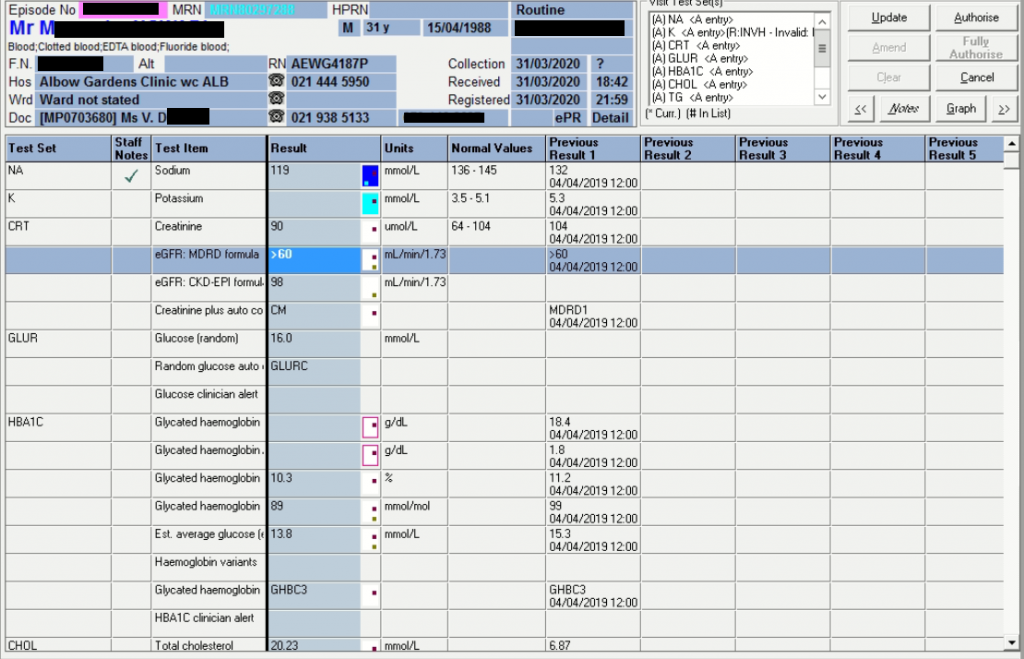
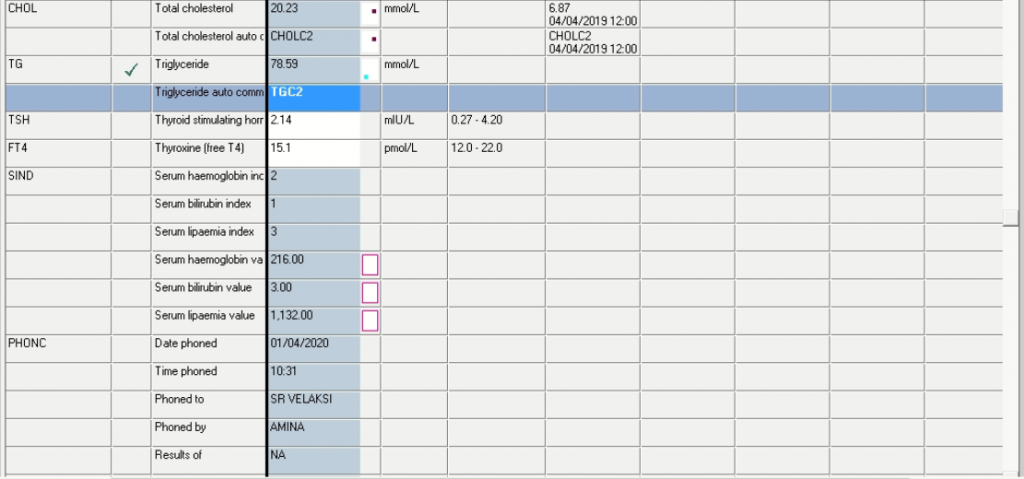
Abnormal Result
An email from a colleague and mentor summarizes the abnormal result the best:
From: Jody Rusch jody.rusch@nhls.ac.za
Date: 2020/05/27 14:26 (GMT+02:00)
Dear S
I am not sure if this will be an easy one to nail down without extensive testing or luck.
To summarise:
Female neonate
No mention of ethnicity
C-section – hydropic on US
Birth weight 2935 g
Low Apgars (2 and 5)
Intubated, ventilated, ICU
Did not grow
Demised on day 7 of life in ICU
Non-immune heart failure plus storage disorder:
IMDs can cause heart failure.
Most likely lysosomal storage disease (14 different ones have been associated with HF)
Most LSDs are AR inheritance
HF, facial dysmorphism, AR inheritance, previous sibling hx
Common in European populations (and it appears globally) include – Mucopolysaccharidosis type VII, Gaucher’s disease, and GM1-gangliosidosis
In SA: Gaucher’s disease (Ashkenazi-Jewish population) – GD2 should be considered in severe perinatal with HF
Extensive list of Lysosomal storage diseases associated with heart failure:
Gaucher disease, type II, Morquio disease, Hurler syndrome, Sly syndrome, Farber disease, GM1 gangliosidosis, I-cell disease, Niemann-Pick disease type A and type C, Infantile Sialic Acid Storage disorder, alpha-neuroaminidase deficiency, multiple sulfatase deficiency, and Wolman disease.
Consider also non-lysosomal diseases
Other IMDs:
Type IV (Anderson disease)
Congenital disorders of glycosylation
Zellweger syndrome
LCHAD
Primary carnitine defic
Smith Lemli Opitz Syndrome
Also hypothyroidism
If a specific diagnosis (beyond likely LSD) is required, and will be paid for, perhaps Invitae have a panel?
Hopefully Prof can weigh in on this and help guide further testing.
Kind regards
J
Presenting Complaint
The histopathologist contacted me regarding any “screening tests” for lysosomal storage diseases
History
Maternal hx:
39 yr old
Booked – normal bloods
35 weeks gestation
Previous pregnancy – stillbirth due to hydrops foetalis (normal karyotype)
No mention of consanguinity
Examination
Post mortem:
Eyes wide set
Left Ear malformed
Flattened nasal bridge
Hydrops foetalis (HF)
Steatosis – lung, liver, heart, placenta
Laboratory Investigations
Not available
Other Investigations
Not available
Final Diagnosis
Unknown
Take Home Message
Message from Prof David Marais:
Hi S & J
Interesting and I wish we could devote much more effort to solve these cases. Especially since this is the second time this mother has had this sad experience and the next pregnancy may result in the same.
On first principles:
- This appears autosomal recessive
- The dysmorphology eliminates many “simpler” inherited errors as homeostasis through the placenta settles imbalances. However, errors involving tissue differentiation, structural components may have dysmorphology. E.g. sterol synthetic defects, mucopolysaccharidoses…It is easy to exclude Smith Lemli Opitz with 1mL serum or plasma even at this stage. However, syndactyly is a very strong feature and hydrops is uncommon but described. Happy to do this if sample is available. Mt abn has been described as well and might explain steatosis though not likely.
- Microvesicular steatosis in several organs is suggestive of incomplete mobilisation of FA into mitochondria for oxidation or inadequate oxidation in mitochondria. These disorders do not usually result in dysmorphology and it is said renal steatosis is typical. LCHAD deficiency has caused hydrops but not dysmorphism to the best of my knowledge. Wolman’s disease has adrenal calcification but not hypoplasia as far as I know and not typically hydrops and diffuse steatosis – will need to check this again. The steatosis could be secondary to severe metabolic stress.
- Hydrops fetalis should be taken as a strong clue. The lysosomal disorders can cause these. The list I found in JIMD Reports (2018) Hurler syndrome (MPS-I; OMIM #607014), Morquio-A (MPSIVA; OMIM #253000), Sly syndrome (MPS-VII; OMIM #253220), galactosialidosis (OMIM #256540), sialidosis
(OMIM #256550), GM1 gangliosidosis (OMIM #230500),
Gaucher type 2 (OMIM #230900), Niemann-Pick disease
types A and C (NPD-A and NPC; OMIM #257200, 257220), Farber granulomatosis (OMIM #228000), Wolman disease (OMIM #278000), mucolipidosis II (I-cell disease; OMIM #252500), sialic acid storage disease (ISSD; OMIM #269920), and multiple sulfatase deficiency (OMIM #272200) which have been shown to be associated.
- Interesting that the spleen is absent and that the adrenals are small. This is hard to explain on any of the metabolic disorders above but I shall have to read more extensively.
- It may be worth testing the urine or other fluids for sialic acid. Infantile Salla disease is a possibility. (Sialin defect, coarse facies, hydrops fetalis, vacuolated lymphocytes but I was not aware of steatosis.) I can look up if this is practicable with the old fashioned assays and chemicals that we do have. I know I tried to analyse sialic acid in the early 1990s. Will look this up too.
One hopes that the anat path dept keeps samples for work-up. Very important to involve the chem path early if any metabolic disease is suspected as one can do fibroblast biopsy up to a few days in the morgue.
Regards
D
Professor Emeritus AD Marais
Chemical Pathology 6.33 Falmouth Building
University of Cape Town Health Sciences
Anzio Rd, Observatory, 7925
Cape Town, South Africa