Patient presented with signs and symptoms of iron deficiency anemia.
History
The patient is a known hemophilia B carrier (Factor IX deficiency or Christmas disease) with menorrhagia and accompanying iron deficiency anemia.
Examination
The patient presented to the emergency rooms with symptoms of severe weakness and had occasional severe menorrhagia.
Unfortunately the physical examination details are not available.
Laboratory Investigations
Iron: 4.5 umol/L (9-30.4) Low
Transferrin 3.92 g/L (2-3.6) High
% Saturation 5% (15 -50) Low
Ferritin 8 ug/L (13- 68) Low
Other Investigations
Anti-Thyroglobulin (anti-TG) antibodies as well as Anti-Thyroid Peroxidase (anti-TPO) antibodies were positive in this patient.
These antibodies was suggested after the results of the TSH and reflexed free-T4 became available and hence after-requested.
Final Diagnosis
The pattern of significantly raised TSH with the significantly low free-T4 and the raised anti-TPO and anti-Thyroglobulin antibodies suggest primary hypothyroidism.
Take Home Message
It’s always worth doing a TSH to screen for thyroid disease when a patient presents with weakness / tiredness, irrespective of the age.
Primary hypothyroidism due to an auto-immune mediated destruction of the thyroid gland tissue is the most common hypothyroid condition and is confirmed by measuring the common anti-thyroid antibodies: Anti-TPO and Anti-TG antibodies. There is likely not much indication for performing these antibodies more than once after diagnosis of hypothyroidism and some clinicians argue it not necessary to even perform these antibody measurements.
Congenital hypothyroidism is one of the congenital disorders causing cretinism which is most preventable by newborn screening. Even though not likely congenital in this patient, it’s worth considering on the differential diagnosis.
The patient was on iron supplements as well as Factor IX injections. I was not aware of an association between Factor IX deficiency and Hypothyroidism, but my Haematology colleagues across the corridor told me the following:
Factor IX deficiency is an X-linked recessive disorder. This makes it unlikely for a female to get this disease. Auto-immune diseases is much more likely in females. There is also a form of Christmas disease where one produces antibodies to factor IX, which yields it inactive, hence presenting as Factor IX deficiency.
This, although unlikely, presents an interesting thought for this unusual presentation in this 16-year old female. Acquired deficiencies of most clotting factors have been described.
However, upon discussion with the attending clinician it seems that the patient did have a clear family history of Christmas disease, hence the presentation.
A case of Cryptococcal meningitis with hypomagnesemia
HOSP #
WARD
Victoria Hospital Female medical ward
CONSULTANT
Heleen Vreede
DOB/AGE
29y female
Abnormal Result
The magnesium result measured 0.36 mmol/L ( 0.63 – 1.05 mmol/L) despite adequate levels prior to admission to hospital (0.75 mmol/L on 18/04/2020).
Presenting Complaint
The patient was asymptomatic with regards to the at the time when the result was obtained.
History
Patient was diagnosed with Cryptococcal Meningitis on 22/04/2020 with a cryptococcal latex agglutination test.
Patient was known HIV positive with a CD4-count of 9 cells/uL (332-1642).
Examination
Unfortunately this data is not available.
The clinical features of hypomagnesemia is predominantly related to the derangement in the calcium becoming deranged when hypomagnesemia occurs.
Laboratory Investigations
Other Investigations
None available.
Final Diagnosis
Hypomagnesemia with accompanying hypocalcemia due to Amphoterecin B therapy
Take Home Message
I’ve learned from the attending clinician (and a short literature search) that Hypomagnesemia is a known consequence of Amphotericin B therapy.
Hypocalcemia is often a consequence of hypomagnesemia (as in this case). This is due to two known mechanisms:
Decreased sensitivity of Calcium at the calcium-sensing receptor, with decreased secretion of PTH and hence its effects.
Decreased action of PTH due to PTH-receptor resistance being caused by hypomagnesemia.
Stata destring with removing weird characters
STATA destring with only keeping numeric numbers
The newest solution to this problem is:
egen-method as described below followed by:
replace problemvar = subinstr(problemvar, ",", ".",.)
https://www.stata.com/statalist/archive/2009-05/msg00862.html
followed by:
destring problemvar, replace *without the 'dpcomma' option.
Dieter Van Der Westhuizen
Mon 2020-07-27 03:01 PM
Sent Items
To:Jody Rusch (jody.rusch@nhls.ac.za);
Follow-up from my previous email:
*This command works when one has either comma or period in the field:
destring kknew3, replace dpcomma
Dieter Van Der Westhuizen
Mon 2020-07-27 02:39 PM
Sent Items
How to strip special characters in STATA variables:
Type the following:
***********************************
program define extrnum
version 7
syntax varlist(max=1) , gen(str)
local maxlen: type `varlist'
local maxlen=substr("`maxlen'",4,.)
tempvar work
qui gen str1 `work'=""
forvalues i=1/`maxlen' {
qui replace `work'=`work'+substr(`varlist',`i',1) if real(substr(`varlist',`i',1))<.
}
gen `gen'=real(`work')
end
*************************************
How to use:
extrnum var1, gen(newvar1)
It works perfectly except it drops the commas too:
**Thus use:
findit egenmore
**and install the app “egenmore” by following the instructions.
**Then one can specify to keep the following characters:
egen kknew3 = sieve(kk), char(0123456789.,)
**Which lets you end up with this:
Destringing this doesn’t work yet, as some characters have “.” and others have “,”
Dieter Van Der Westhuizen
Mon 2020-07-27 11:30 AM
Sent Items
Dieter van der Westhuizen Chemical Pathology RegistrarC17 NHLS Pathology Laboratory, Groote Schuur Hospital and Red Cross Children’s Hospital LaboratoryTel: 021 404 4135 | Cell: +27 82 861 2093 | Fax-to-email: 0866 090 397dieter.vdwesthuizen@nhls.ac.za | www.nhls.ac.za
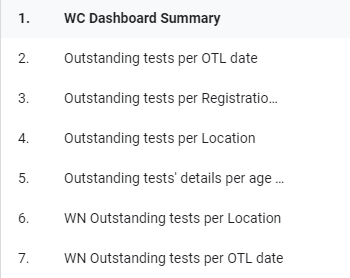
Section 7.8 – COVID OTL Dashboard
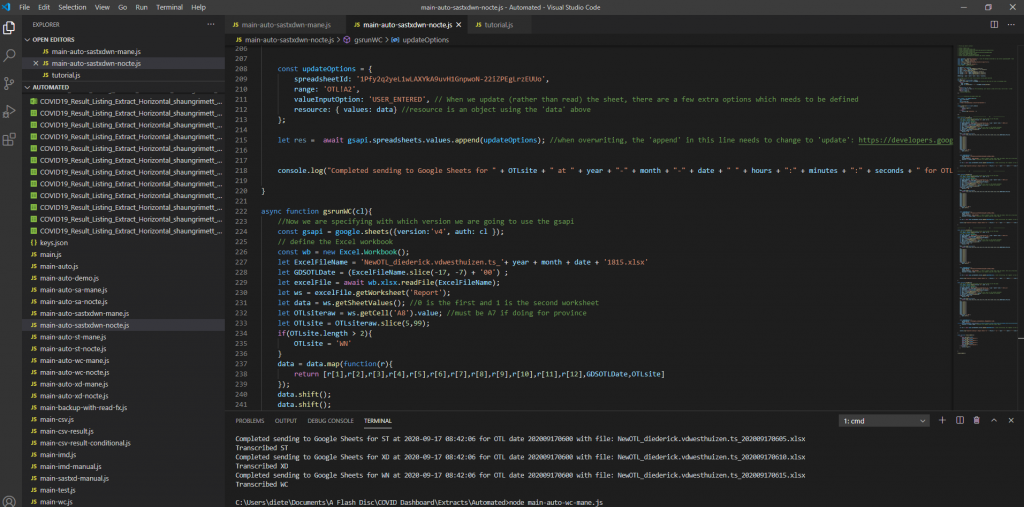
A dashboard was created to visually represent the COVID outstanding test list for our province for COVID outstanding PCR tests. This dashboard has been requested by the area manager of the Western Cape and various others on the Virology Expert Committee. Even though not a task primarily assigned to chemical pathologists, since I have an interest in data science, I tried to help. The end result was a dashboard which is updated every morning at 06h00 and every evening at 18h00 with a few JavaScript scripts running each day, updating three databases on the backend along with automated data extractions being done from TrakCare every morning and evening at a predefined time.
Screenshot 1 – Illustration of the JavaScript code to read the extracted Excel (or CSV) files and transcribing them to a Google Sheets database.
This dashboard was used (and likely still are being used) especially by the virologists at Groote Schuur Hospital to track the progress of outstanding COVID PCR tests and it can also be used to show possible bottlenecks in pre-analytical sample issues if tests are already registered before being sent to any of our laboratories in the Western Cape.
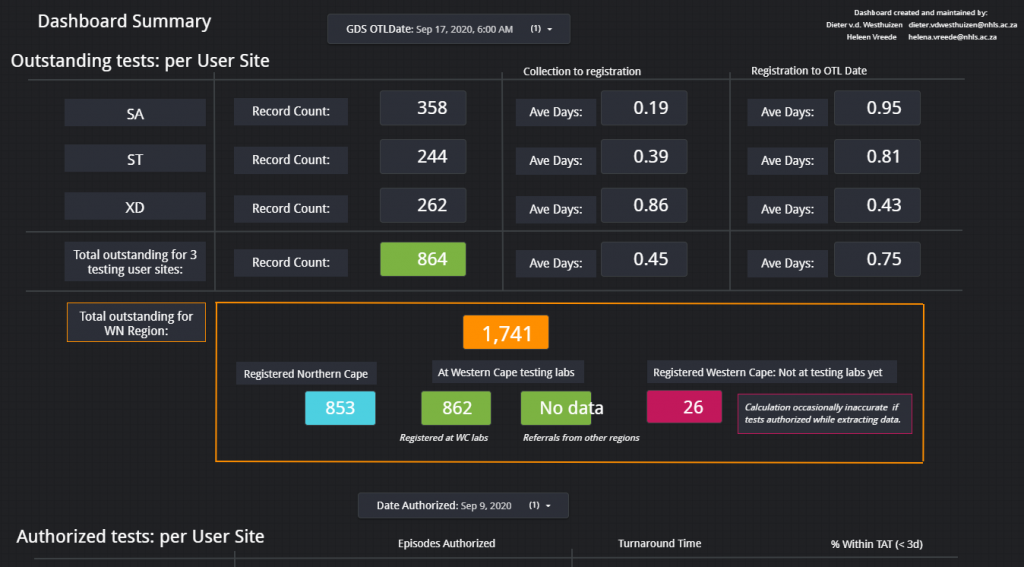
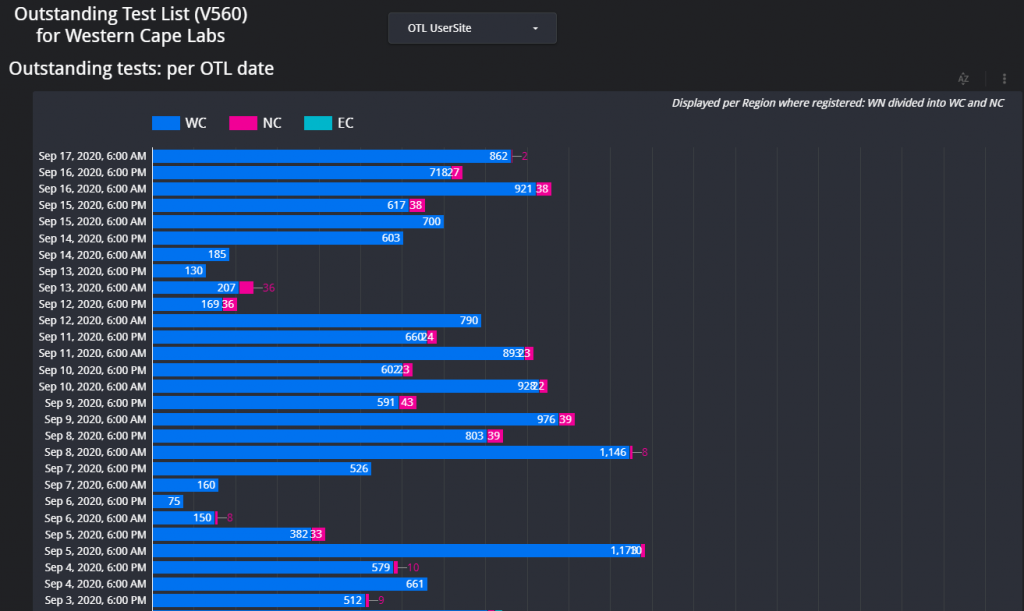
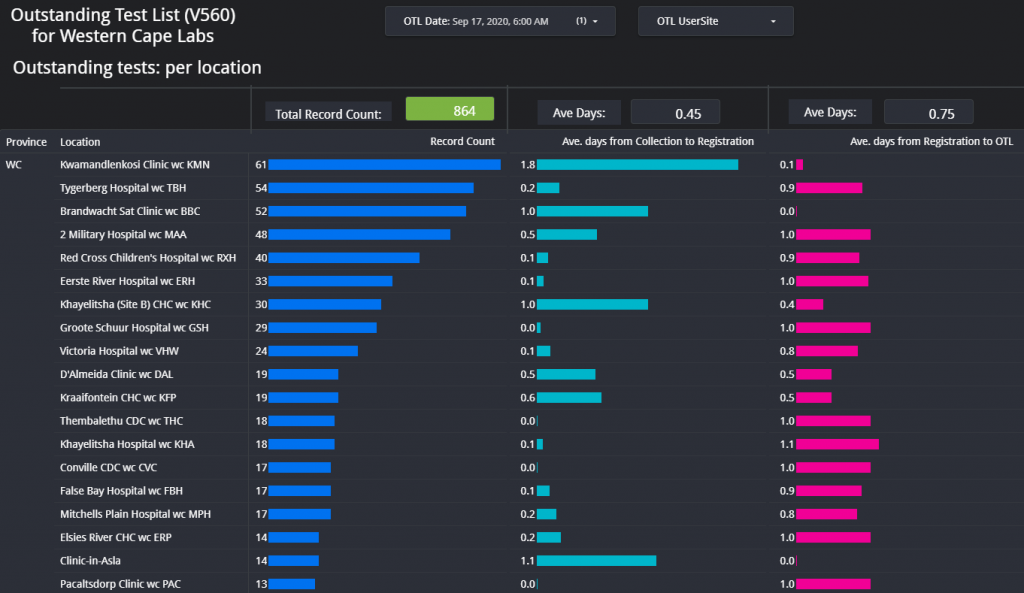
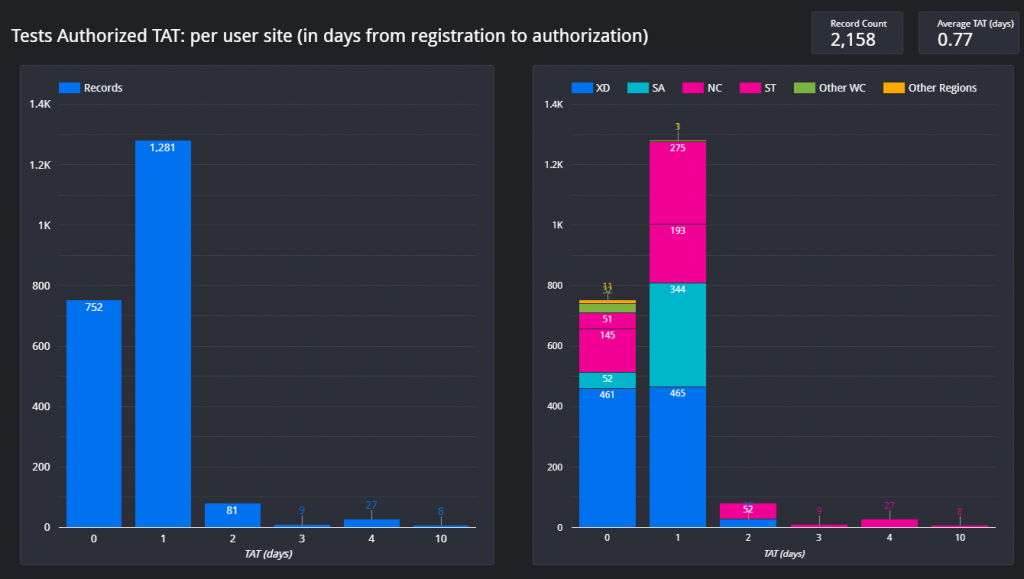
Figure 1 – Screenshot of the Dashboard. Follow the link above to view.Figure 2 – Example of the layout of the page which shows the total count of items on the respective outstanding test lists for each respective data for the Western Cape.Figure 3 – Outstanding tests summary by location. This page is especially helpful if the delay / outstanding tests from a specific hospital or clinic needs to be visualized in comparison with other locations (hospitals / clinics).Figure 4 – Illustration of the turnaround time met by a certain count of samples per each respective user site for one day.
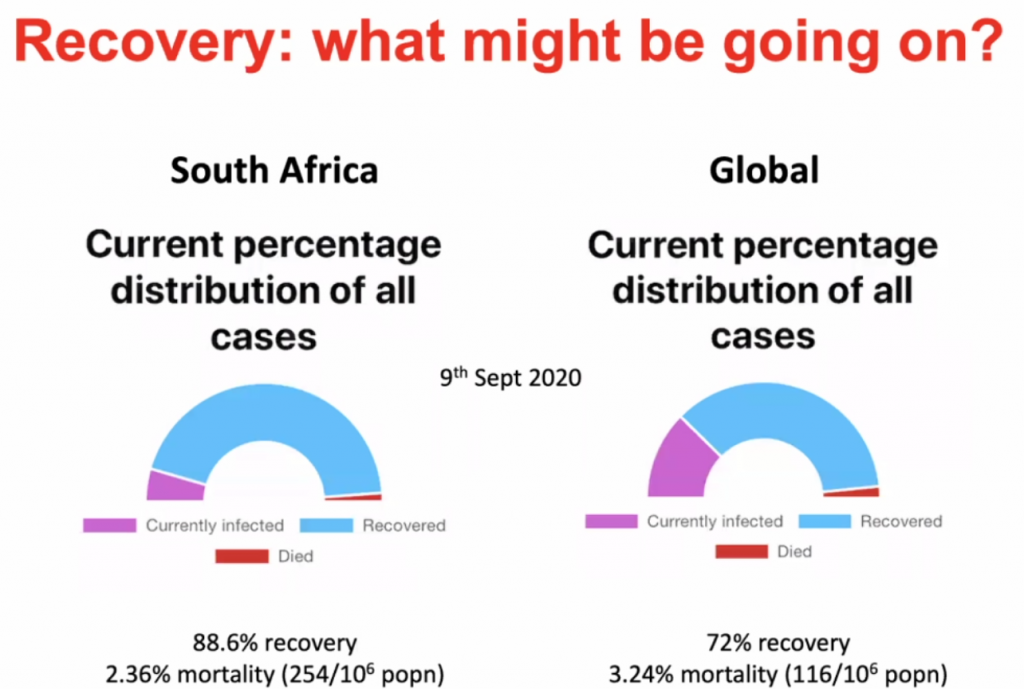
Immune Responses to SARS-CoV-2 cause severe COVID-19 in some and recovery in most
Clive Gray – Professor in Immunology immunopaedia.org – useful web site for immunology resources.
Outline:
Basics of Immunology
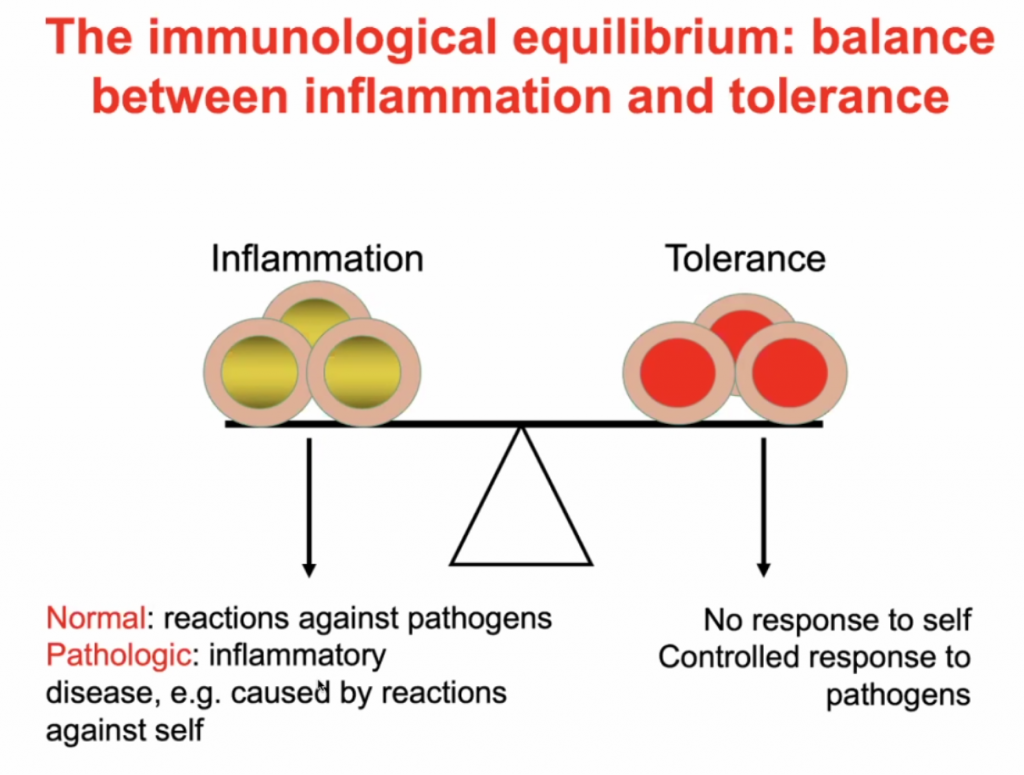
A balance between inflammation and tolerance
What happens to people who progress to severe COVID-19?
What might be happening in SARS-CoV-2 infected people who remain asymtomatic, have few symptoms and recover?
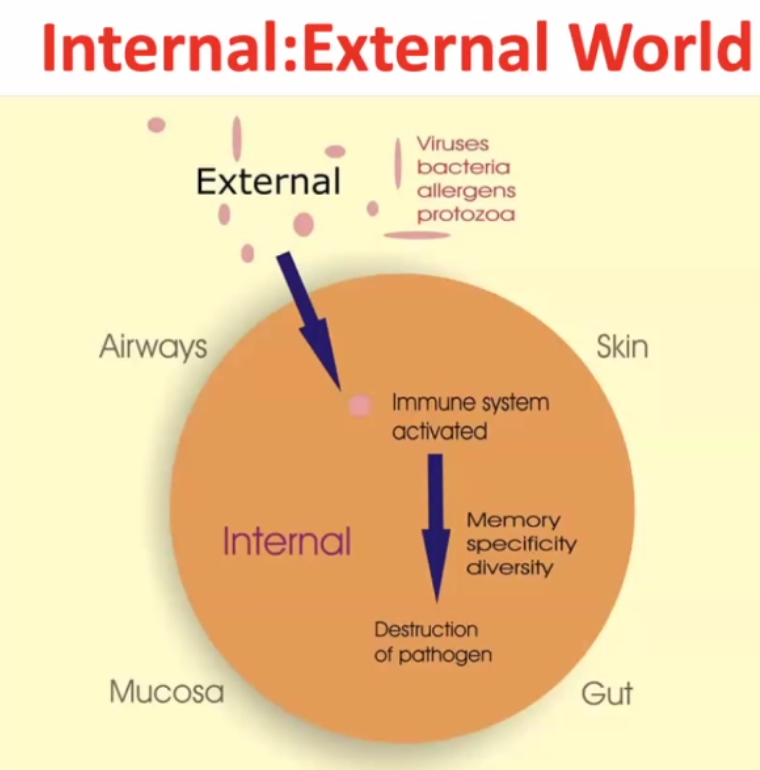
Internal : External world
~99% of time the pathogen gets destructed, but the pathogen may survive in rare cases.
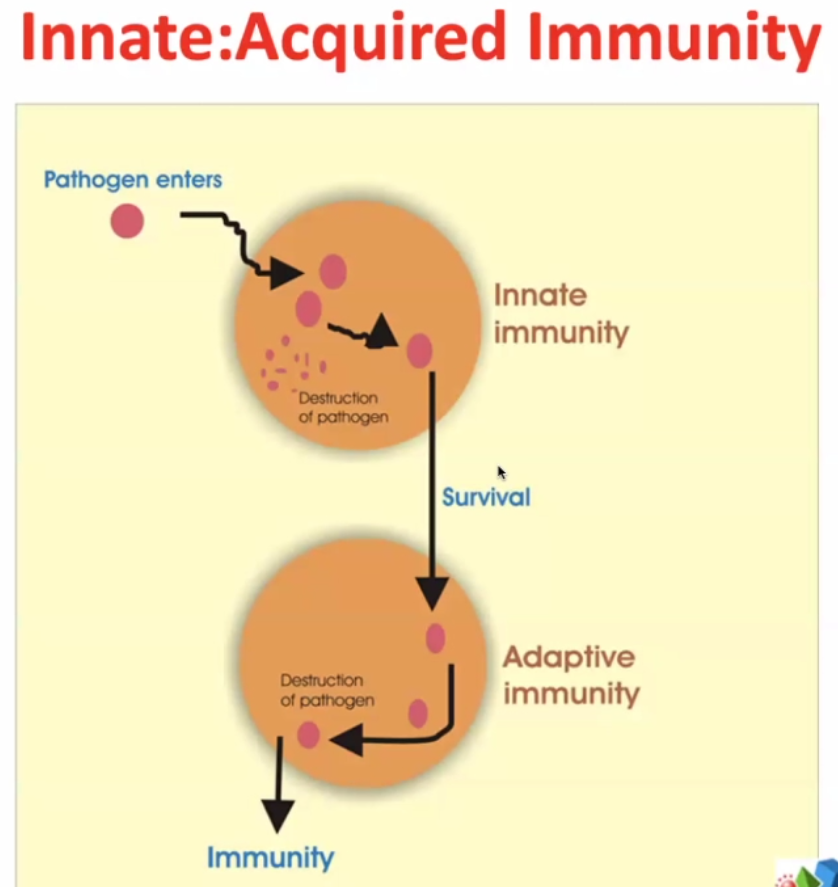
2 arms of immune responses:
Innate – evolutionary response – very rapid – elements of innate immunity are found in bacteria, plants, lower vertebrates, squids, fish etc.
Some pathogens survive ->
Adaptive immunity – much more targeted / focussed. The immune system targets more specifically the pathogens which survive the innate immunity.
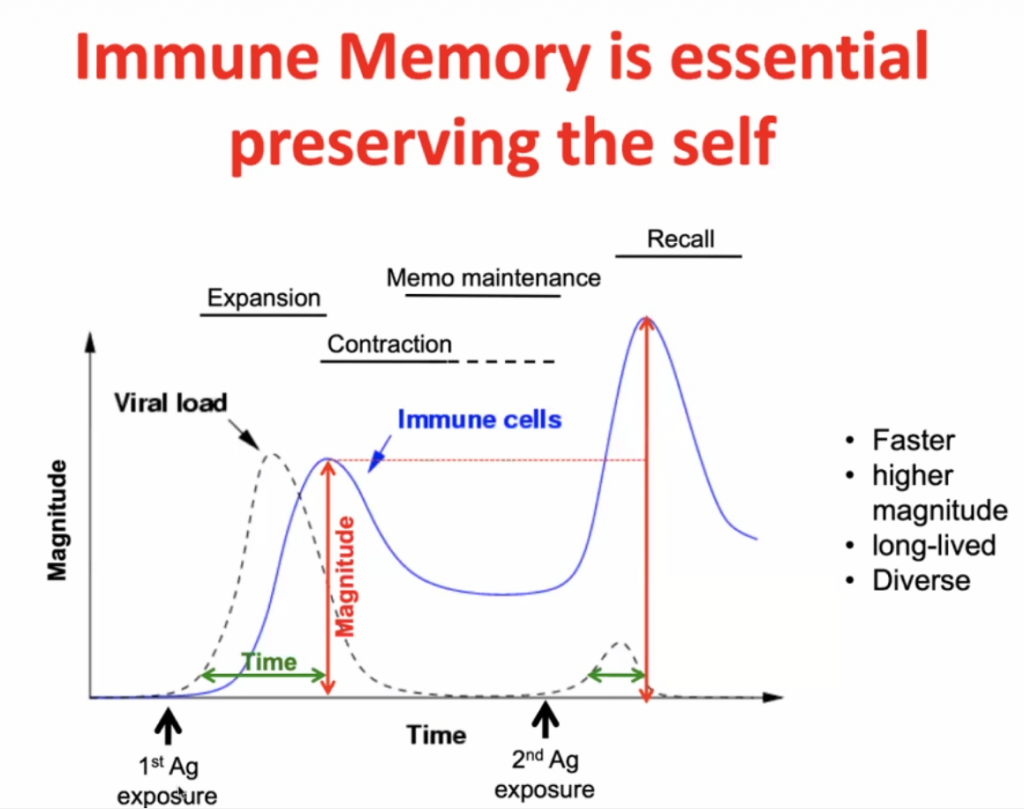
Infection initially -> expansion (peak after maximal viral load) -> contraction with some residual immunity (Memo Maintenance) -> with secondary response (Recall) there is a more rapid expansion (and higher peak) of the specific immunity.
Immune regulation:
Predisposed conditions: DM, HPT, Obesity, would make an individual highly susceptible to inflammation due to in imbalance of Inflammation vs. Tolerance, see below.
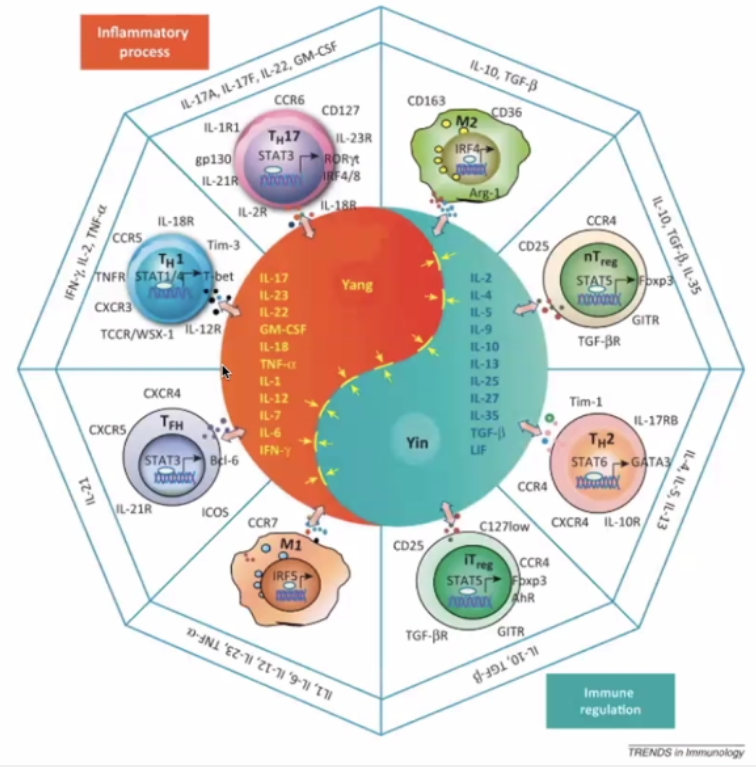
The Yin Yang of immunology:
Yin – immune regulation; Yang – Inflammatory Process
Pro-inflammatory (Orange)
TH17 – inflammatory cells secreting the “calling signals” for leucocytes.
Macrophage – presents antigens – in lymph nodes and germinal centres
T-Helper cells T-FH
Immune Regulation (Blue)
TH2 – hand in hand with TH1 (opposite)
Regulatory cells (nT and iT regulatory cells)
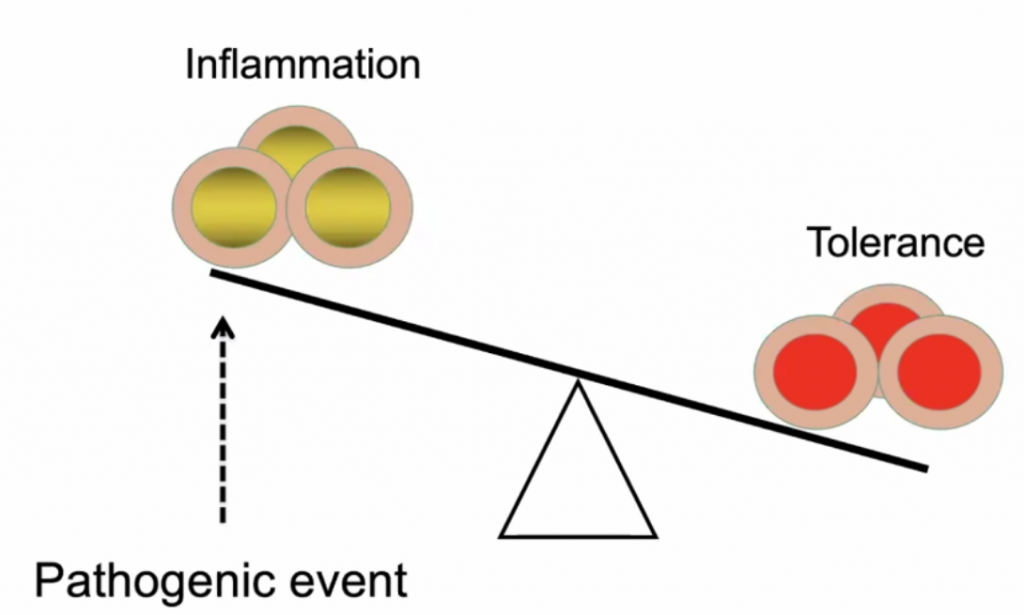
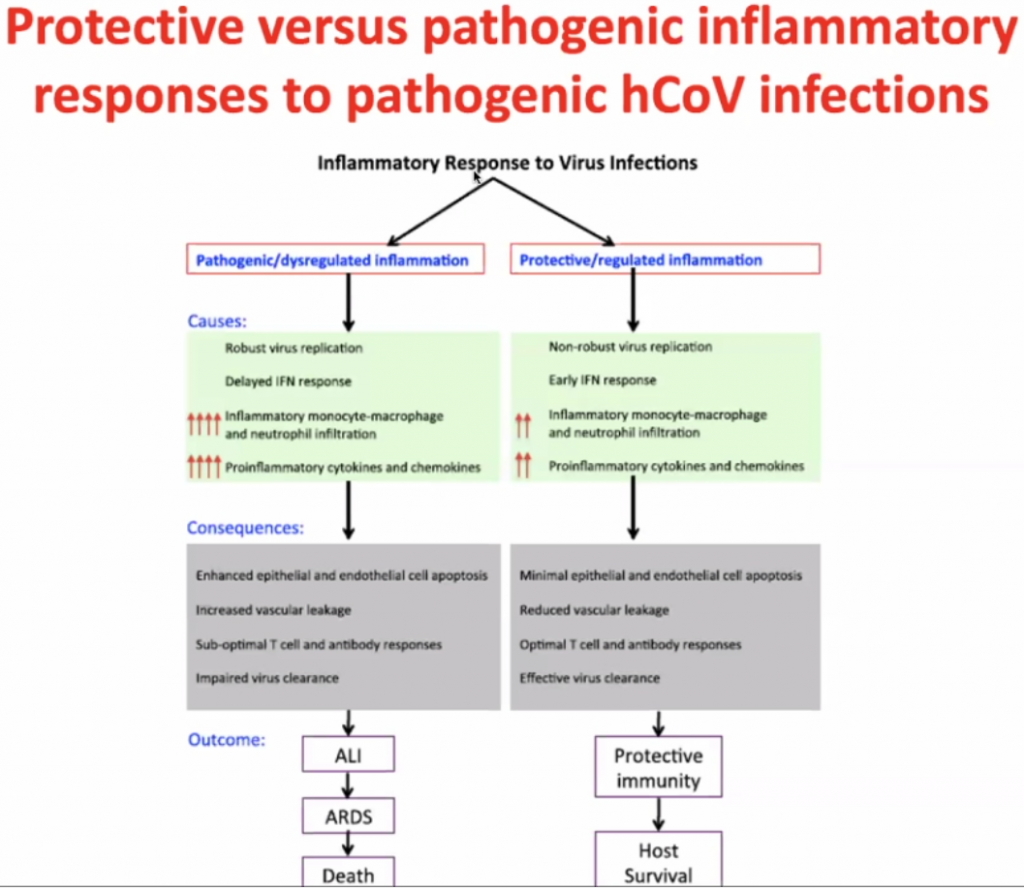
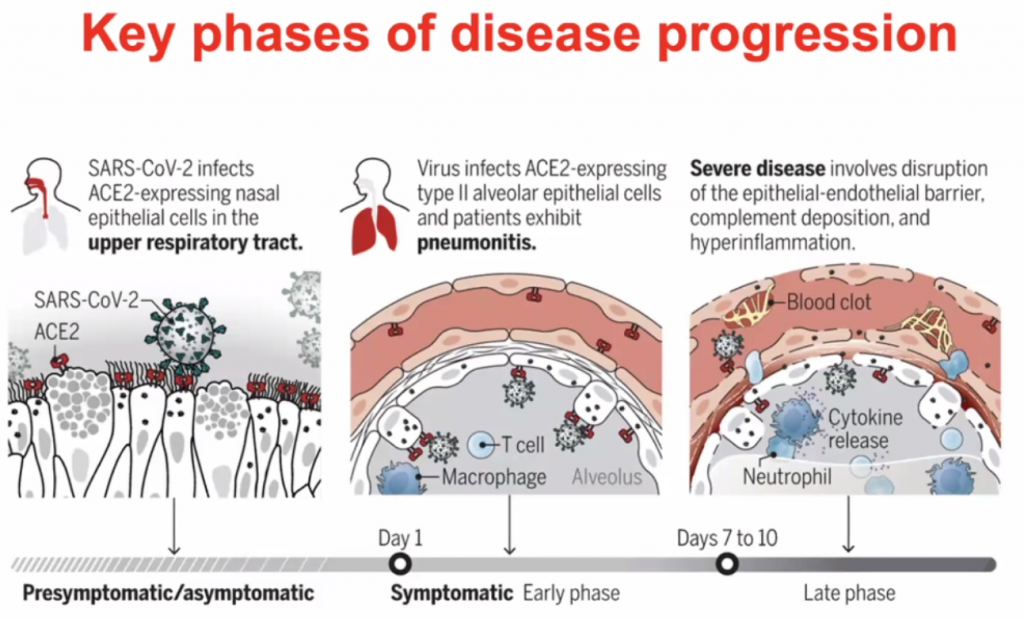
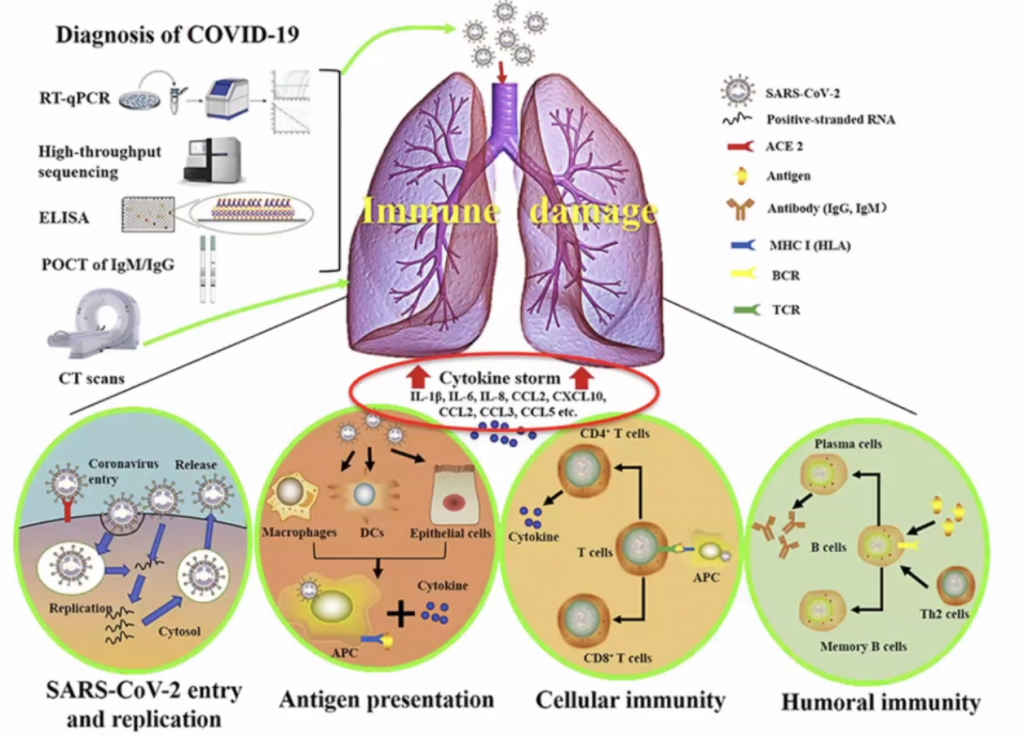
Actual pathogen is not causing disease – but the immune response – thus this is what should be focussed on to treat the disease.
Dose of the virus (viral load) is key to how you respond to the virus – Initial High dose in viral load likely will lead to high inflammatory response; Low dose (non-robust virus replication) may cause a less severe inflammatory response.
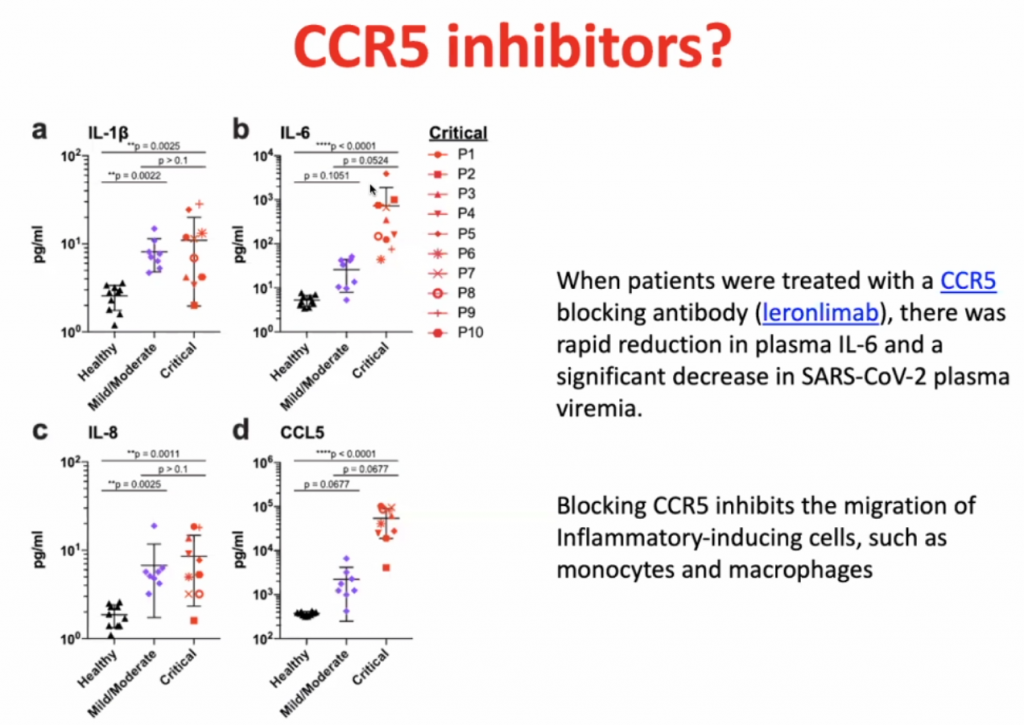
CCL’s allow leucocytes to migrate, hence in a cytokine storm, with high level of migration, the leucocytes causes severe local inflammation due to migration of leucocytes to local sites.
CCL5 blocking antibodies leads to rapid reduction of IL-6.
Dexamethasone is not so much an inhibitor of CCR5, but it prevents the hyperinflammation by inhibiting the majority of the inflammatory pathway.
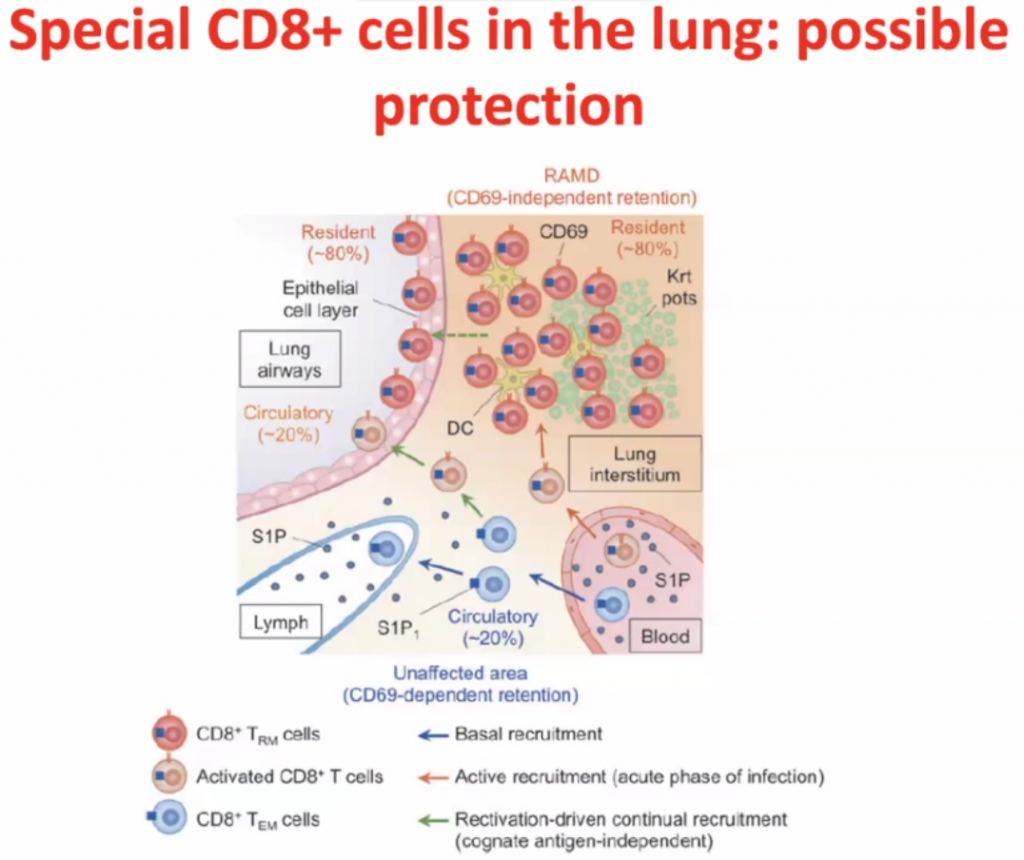
CD8 cells
Within interstisium, the CD8 cells are present and
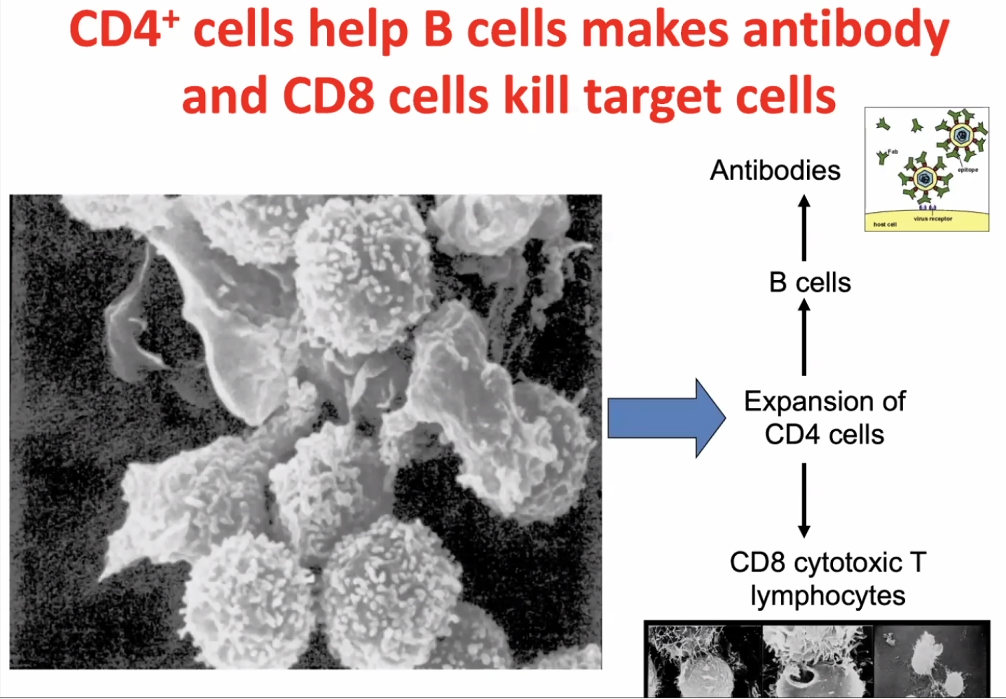
CD4 cells activates CD8 cells, hence called T-helper cells.
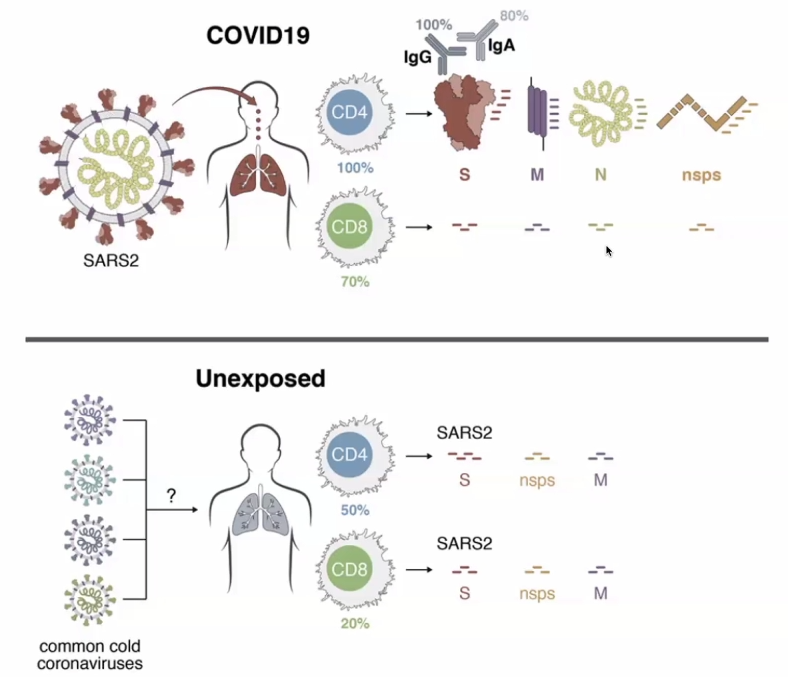
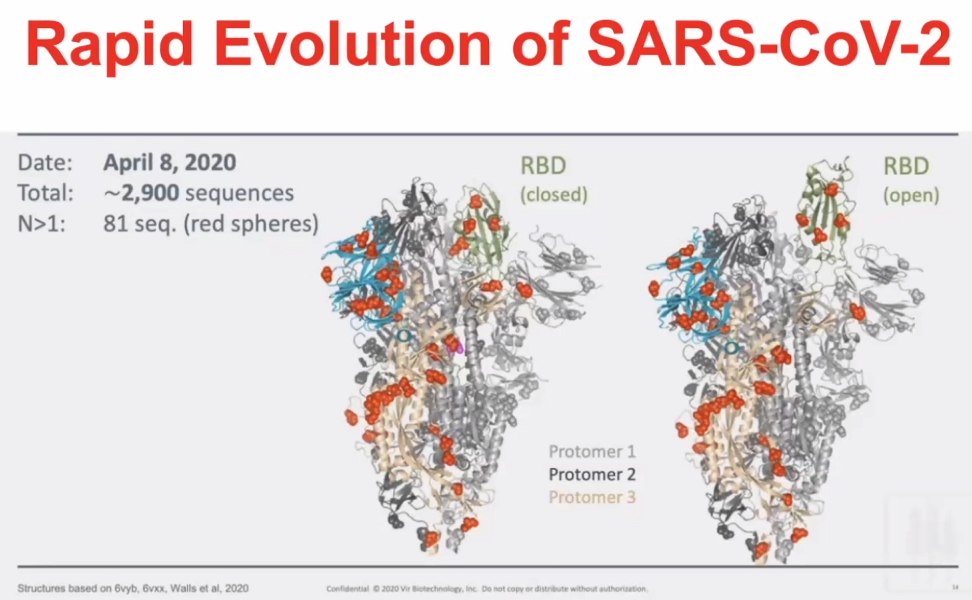
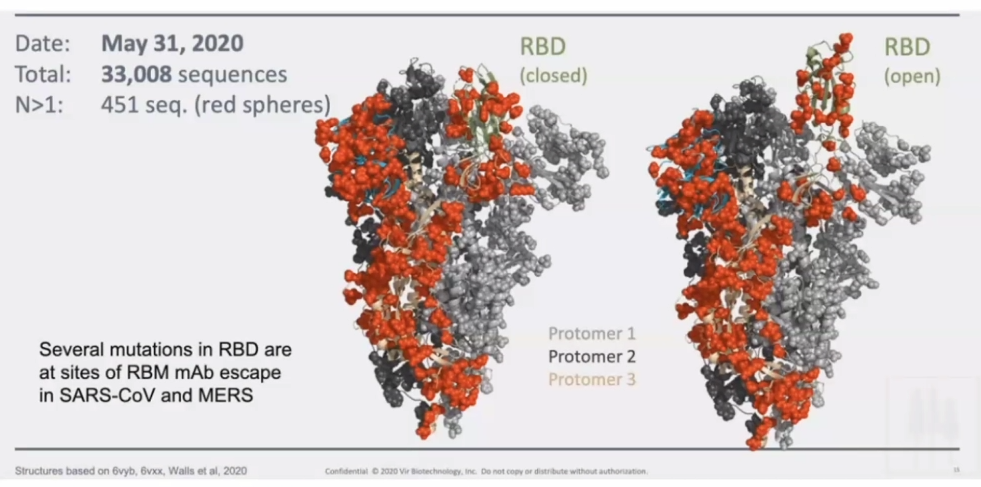
T-cell responses are very prevalent to COVID-19 exposed individuals. BUT CD4 cells and CD8 cells can also react to the SARS2 viral proteins in unexposed individuals.RBD – receptor binding domain (Spike-protein); Orange is the amount of amino acids which are changeable.Orange amino acids are those which are changeable – illustrating how the virus has mutated in a month.
A pepper-pot skull?
HOSP #
WARD
General Practitioner Practice in Robertson
CONSULTANT
Dr. Jody Rusch
DOB/AGE
83 year Male
Abnormal Result
Serum protein electrophoresis demonstrates a 4.4 g/L, IgG kappa monoclonal peak in the gamma region.
Presenting Complaint
Complains of bilateral hip pain and RUQ discomfort.
History
Atrial fibrillation on Xarelto.
2 x CABG
Examination
RUQ pain and tenderness
Hear rate regular
Laboratory Investigations
Urine protein electrophoresis: No Bence Jones protein
Serum free light chains:·
Kappa 62.87 mg/L (3.30-19.40)·
Lambda 19.63 (5.71-26.30)·
K:L ratio 3.20 (0.26-1.65)
Creatinine 108 (eGFR 56)
Calcium 2.42 mmol/l
Albumin 40 g/L
Hb 12.7 (11.0-16.0)
Other Investigations
U/S shows gallstones.
X-Ray of pelvis shows “sclerotic changes to both hips and pelvis”
Final Diagnosis and Take Home Message:
1. What is the likely diagnosis
This 83 year old male with multiple co-morbidities presenting with signs and symptoms suggestive of multiple myeloma, confirmed on SPE as IgG Kappa.
CRAB criteria before performing SPE: C- R+ A- B+ (2/4)
Bony pain could be secondary to lytic bone lesions associated with MM, but also possibly to due sclerotic/ wear-and-tear when considering his age. RUQ pain is likely due to gallstones.
Renal impairment – this is probably normal renal function for an 83 year old man
In medicine generally an eGFR < 60 is representing renal impairment (stage 3)
However, in monoclonal disease eGFR < 40 or serum Cr > 177 is the cut-point
Bone lesions – myeloma classically causes lytic bone lesions, e.g. “pepper-pot skull”
It was suggested that the clinician talks to the radiologist as to whether the X-Rays were in keeping with myeloma.
2. Critically discuss whether this patient needs a bone marrow biopsy.
The patient’s age along with co-morbidities would concern any drastic intervention:
he will be an anesthetic risk for BM Bx to be performed in theatre (assuming that is standard procedure), and
will the BM biopsy give add anything further to the already established IgG Kappa diagnosis, which can be treated accordingly.
The case should ideally be discussed with Oncology. A bone marrow biopsy is done under local anaesthetic. The bone marrow will allow the haematologist / oncologist to assess the degree of marrow clonal infiltration. The important cut-offs are 10 & 60%. This is important to decide on diagnosis, stage, prognosis, treatment and later, the response to treatment. The criteria for doing a bone marrow biopsy at our centre are:
Positive CRAB·
IgG monoclonal peak > 15 g/L·
IgM or IgA monoclonal·
FLC K:L > 10
Why is there a lower (10%) limit for degree of marrow clonal infiltration? Is there a link to immunoparesis? One likely always has some clonal expansion in bone marrow, probably a normal or a non-pathological finding.
3. Discuss the serum FLC in the setting of the renal impairment.
FLC are filtered and reabsorbed by the nephron under normal circumstances, along with other LMW proteins. During a plasma cell dyscrasia, the nephron is overwhelmed by the amount of FLC (stemming from monoclonal origin), can cause renal impairment. Hence, renal function being part of the CRAB criteria. Furthermore, renal impairment itself (in the absence of MM), can cause elevated Kappa and Lambda FLC – usually with a slight higher ratio =3.2.
In patients with renal failure, there is greater retention of serum free light chains. It is difficult to interpret ratios ranging between 1.65 – 3.0 in the context of renal insufficiency. In such cases, further investigation with a 24-hour urine protein electrophoresis and urine immunofixation helps to guide interpretation. If both of these subsequent studies are normal and the patient has no other symptoms suggestive of a plasma cell dyscrasia, then the increased ratio is likely due to the renal insufficiency.
4. Discuss electrophoresis briefly.
Electrophoresis is a general term that describes the separation of charged particles/ ions under the influence of an electric field – in this case the charge of proteins. Migration of proteins is based on their charge, size and velocity (product of their mobility and field strength) Make sure you understand why the proteins are charged the importance of NET charge and how we keep those charges stable in the field. If I can take a crack at this: The overall NET charge of the molecule is based on the number of elements (incl. amino acids with varying side-chains moeities) (I think this is the confusion when some mention that electrophoresis is based on charge, and also size. I don’t necessarily think that the two are synonymous), and each amino acid has different degrees of charge based on their differing R-group. The stability of the charges within the field is achieved by running the sample solution through a buffer. Right about the buffer. Remember that size and charge are two different physical aspects that you can use to separate molecules. For example, a DNA gel is a separation purely based on size. The net charge is the same on all the molecules. The net charge in proteins is from the side chains, which is why you have to learn about neutral, acidic and basic amino acids. The side chains have different pKa’s and so are charged differently.
a. What is the difference between capillary and gel electrophoresis. Explain which your lab uses and why.
What I described in Q4 was basically the concept of gel electrophoresis where agarose gel is used as the medium in which the proteins are separated according to their size, charge, and interaction with the medium itself. At TBH we use gel electrophoresis, but will soon be getting a Minicap/ CZE. CZE: As with gel electrophoresis, CZE also separates ions based on their electrophoretic mobility with the use of an applied voltage – all dependent on the charge of the molecule, viscosity and particle size. CZE’s voltage is much higher compared to GE – quicker results. The buffer/ mobile phase of CZE uses an electrolyte filled capillary, where eletro-osmotic flow (EOF) is generated: similarly sized and charged ions move together and are subsequently separated and detected at different time intervals.The more voltage you apply the faster the separation occurs. However, the limiting factor is that applying high voltages generates a lot of heat which can denature proteins, thereby altering their conformational shape and changing their NET charge. Capillaries are much more effective at shedding heat as they are long and thin. Thus, very high voltages can be applied and the run time is much shorter. In gel electrophoresis, you measure how far the molecules travel in a set time, e.g. 1 hour. In capillary electrophoresis, the distance that the molecules move is set and so you measure the time it takes for the molecules to travel that set distance (like running a 100m race).
The way I reconcile how the CZE differential seperation works is by the
driving force of the buffer through the tubing (forward force)
negative charge on the side of the tube (retarding force)
NET charge on the molecule (many amino acids=higher charge, eg Albumin) (determine degree of retardation of flow)
Voltage powers the whole system
5. Why is the serum FLC abnormal but not the urine protein electrophoresis?
UPE’s sensitivity is limited due to the reabsorption of FLC in the renal tubules. FLC in urine will only be detected until loss of tubular function/ tubules are overwhelmed by FLC volumes. This patient’s Kappa FLC of 63 mg/L in serum should be detected on UPE, but tubular function is seemingly still intact with little being excreted.
Some are of the opinion that SPE and SFLC is the preferred method to screen for myeloma because of higher sensitivity and specificity, as opposed to SPE and UPE, which may have a slightly lower sensitivity.
It should however be noted that quoted sensitivities and specificities are usually based on retrospective audits of patients who eventually end up in a myeloma clinic. So, it is not sure what the sensitivity and specificity is if you just screen the general population, older people, people with some vague symptoms…
6. Against which epitopes are the FLC assay directed?
The FLC epitopes are located between the interface between the light and heavy chains and are “hidden” – when bound to Ig, they will not be detected. Only when these epitopes are “free”, can they be detected, hence free light chains. They are directed at 2 hidden epitopes.
7. Why is the FLC assay polyclonal and not monoclonal?
The biggest decider many times is COST, but lets put that aside for now.
It appears that polyclonal assays are more robust and have higher yields in product during testing and easier to make. They are unfortunately less specific, but this is not the most critical when one wants to measure the FLC broadly, instead of particlularly specific sites.
Epitopes are three dimensional shapes that the antibody binds to. This is determined by the amino acid sequence. One drawback with polyclonal assays is that lot to lot will vary. The difficulty is to maintain consistency in further production and / or distrubution of the antibody – it is not a simple process to ensure consistency.
8. Describe how a monoclonal antibody is made for use in an assay.
Inject a rabbit (or other animal) with the protein of choice. In three weeks, the rabbit will have produced antibodies to the protein. The rabbit is sacrificed (killed) and the spleen harvested. The spleen is ground up and the cells are put in a culture with a certain myeloma cell line. The culture medium contains colchicine that induces the rabbit cells and the myeloma cells to fuse. It also contains HAT medium: hypoxanthine, aminopterin and thymidine. This specific myeloma cell line cannot recycle thymidine in the presence of hypoxanthine.
So in the culture there are now three cell lines.
Firstly, there are the rabbit cells that haven’t fused; these will die because they are not immortal.
Secondly, there is the myeloma cell line, this will also die because of the recycling problem.
Finally, there is a fused cell line that will survive .
Each of these surviving cell lines will produce one Ig against one part of the protein. Now the researchers take the medium and put a tiny amount into a well. The amount is so small that on average each well will contain only one cell; some will of course contain nothing. Then, each well is targeted against the protein and the most promising ones are investigated further. An immortal Ig producing factory directed against one epitope and based on one cell line, a single clone, or as we’d call it a monoclonal, has been produced. Each manufacturer’s produced immunoglobulin is different and may produce better or worse results.
9. The GP, in Robertson, wants some advice on how to proceed. What do you tell him?
A multidisciplinary approach would be best:
Treatment for the lytic bone lesions (after opinion by radiology): bisphosphonate
Assess overall medication and lifestyle to determine overall risk for worsening renal dysfunction (drugs, co-morbidities……always suggest stopping smoking/drinking)
Prevention of thrombotic/infective episodes
Treatment of any further abnormalities should they arise (hypercalcaemia, anaemia etc.)
Specialist referral:
Haematoncologist for treatment of MM: UPE , Bone marrow biopsy
General surgery for gallstone
10. Is there any relevance for the RUQ/gallstone pain in myeloma specifically?
There are some reports where cholecystitis has presented in MM (mets etc), but it is not a separate entity on its own (such as in POEMS), this is simply the real world where elderly patients have more than one pathology.
Excessive amino acids in the Urine
HOSP #
WARD
A1 Paediatric Ward – Grey’s Hospital
CONSULTANT
Prof. George van der Watt
DOB/AGE
27 day old Female neonate
Abnormal Result
Sodium 186mmol/L
Presenting Complaint
The neonate was taken to the Emergency Department due to seizures.
History
Unfortunately the attending clinician at Greys hospital did not have much of the history.
There was no history of diarrhoea according to what she remembered.
Examination
Patient was severely dehydrated clinically.
Later the patient presented with edema, signs and symptoms of nephropathy and biochemical changes in keeping with liver failure.
Laboratory Investigations
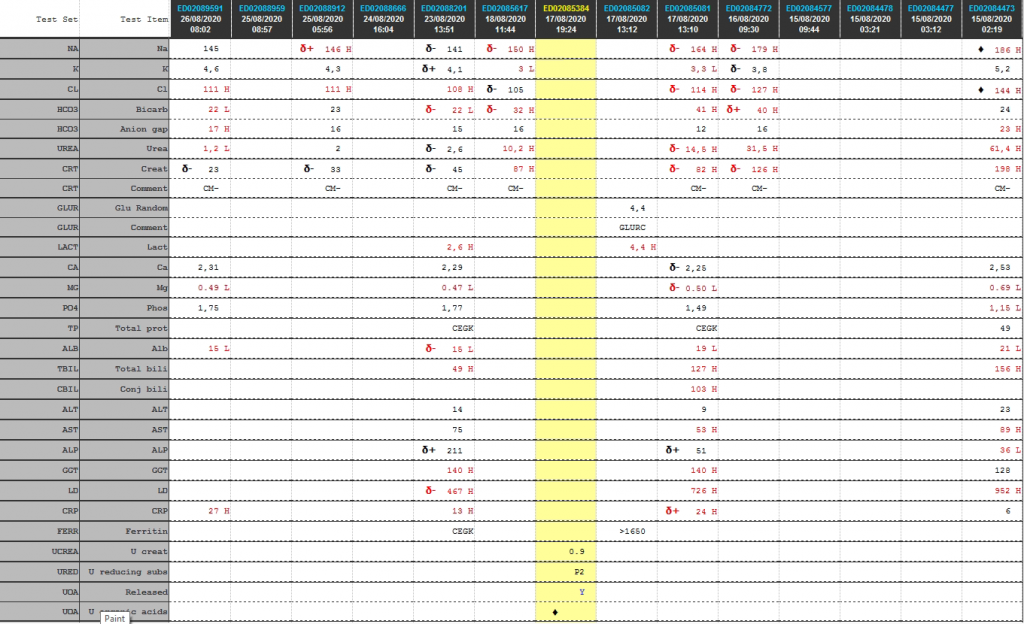
Cumulative laboratory history
Test
26/08/2020
25/08/2020
23/08/2020
18/08/2020
17/08/2020
16/08/2020
15/08/2020
Na
145
δ+ 146 H
δ- 141
δ- 150 H
δ- 164 H
δ- 179 H
♦ 186 H
K
4,6
4,3
δ+ 4,1
3 L
3,3 L
δ- 3,8
5,2
Cl
111 H
111 H
108 H
δ- 105
δ- 114 H
δ- 127 H
♦ 144 H
Bicarb
22 L
23
δ- 22 L
δ- 32 H
41 H
δ+ 40 H
24
Anion gap
17 H
16
15
16
12
16
23 H
Urea
1,2 L
2
δ- 2,6
10,2 H
δ- 14,5 H
31,5 H
61,4 H
Creat
δ- 23
δ- 33
δ- 45
87 H
δ- 82 H
δ- 126 H
198 H
Glu Random
4,4
Lactate
2,6 H
4,4 H
Ca
2,31
2,29
δ- 2,25
2,53
Mg
0.49 L
0.47 L
δ- 0.50 L
0.69 L
Phos
1,75
1,77
1,49
1,15 L
Total prot
CEGK
CEGK
49
Alb
15 L
δ- 15 L
19 L
21 L
Total bili
49 H
127 H
156 H
Conj bili
103 H
ALT
14
9
23
AST
75
53 H
89 H
ALP
δ+ 211
δ+ 51
36 L
GGT
140 H
140 H
128
LD
δ- 467 H
726 H
952 H
CRP
27 H
13 H
δ+ 24 H
6
Ferritin
CEGK
>1650
Table 1. Cumulative laboratory results history from newest to oldest
Interpretation:
Urine organic acid analysis by GCMS demonstrates elevation of the liver markers
4-OH phenyllactate and 4-OH-phenylpyruvate together with lactaturia.
Succinylacetone, a marker for tyrosinaemia type 1 is absent. These changes
indicate underlying hepatic dysfunction with lactataemia but are non-specific
for an IMD per se. Rare forms of lactataemia include defects in pyruvate
metabolism (gluconeogenic defects such as glycogen storage disease type 1,
pyruvate dehydrogenase deficiency and thiamine deficiency). In these disorders
the lactate/pyruvate ratio is normal despite lactataemia. In pyruvate
dehydrogenase deficiency the CSF/Plasma lactate ratio is typically >2. Please
note that routine metabolic screening does not exclude galactosaemia. If
galactosaemia is suspected this should be screened for by measuring red cell
GALT activity in unspun heparin whole blood or by screening for the common
African S135L mutation in Black South African patients. As part of our
gatekeeping policy to limit unnecessary testing, routine metabolic screening at
Red Cross Children's Hospital consists only of urine organic and amino acid
analysis. Additional tests must be requested separately based on the working
differential diagnosis and routine screening results. Relevant legible clinical
information also aids significantly in interpreting metabolic profiles.
Other Investigations
Urine dipstick: Blood 3+ Protein: Trace Glucose (by glucose oxidase) ++/+++ 28 – 55 mmol/L (hence this is most likely the predominant reducing sugar – as suggested by Prof David). This is an interesting finding since the random glucose in the ward the same day was 4.4 mmol/L. Hence two possible theories: likely either severe tubular injury or, when the child had convulsions they gave a dextrose infusion which increased the plasma glucose above the renal threshold. pH >8.5 Nitrites: Not present Leucocytes: Not present Urobilirubin: Trace Bilirubin: Not present
Final Diagnosis
Severe dehydration with acute kidney injury (pre-renal origin) is the most likely cause of the presentation of seizures.
Take Home Message
Dehydration is a common cause of pre-renal acute kidney injury
My thoughts initially, was that the urine amino acid screening by GCMS demonstrated a heavy generalized aminoaciduria indicative of renal tubulopathy and that cystinosis should be considered as this is the most common inheritable cause of renal tubulopathy in South Africa. However, this was later decided to be changed to rather and “evolving tubulopathy” clinical picture, as shown in the two examples below.
Typical Urine Amino acid profiles of a patient with confirmed cystinuria (left) and a patient with tubulopathy (right)
Fanconi syndrome — Generalized proximal tubular dysfunction, referred to as Fanconi syndrome, is characterized by phosphaturia, renal glucosuria (glucosuria with a normal plasma glucose concentration), aminoaciduria, tubular proteinuria, and proximal RTA.
The etiology of Fanconi syndrome includes inherited diseases or acquired causes [Source: Up-to-date]:
Genetic conditions associated with Fanconi syndrome include the following:
Dent disease (X-linked recessive nephrolithiasis)
Cystinosis
Tyrosinemia type 1
Galactosemia
Wilson disease
Lowe oculocerebrorenal syndrome, also referred to as Lowe syndrome
Hereditary fructose intolerance
Mitochondrial myopathies
Acquired causes of Fanconi syndrome include:
Drugs – Medication associated with Fanconi syndrome include
aminoglycosides
cisplatin
ifosfamide
valproic acid
deferasirox
Heavy metals
lead
mercury
cadmium
The fact that the urine dipstick was positive for glucose, suggests that either the acute kidney injury is the source of glucosuria or the patient was treated with high dose dextrose, causing the plasma glucose to overwhelm the tubular threshold for glucose transport.
Hemochromatosis
How to remember the side effects of hemochromatosis
Renal tubular acidosis
Two one four, low low more.
A case of raised PSA with ALP
HOSP #
Lab no. SA04016354
WARD
Orthopaedic Clinic
CONSULTANT
Jody Rusch
DOB/AGE
61y Male
Abnormal Result
PSA: 846.5 ug/L
ALP: 284 U/L (53 – 128)
Presenting Complaint
Painful ”lumps” in groin + constipation
Spine pain
History
Smoker (>45 years)
No other co-morbidities
6/12 history of generalized body pain (mostly spine)
Red Flags (weightloss, night pain not responding to analgesia)
Examination
O/E: Pallor (Hb 8.6), Wasted. Clinically painful bilateral inguinal lymph nodes PR: normal tone, no masses, no blood, prostate smooth
Laboratory Investigations
Na
138 mM
K
4,7 mM
Cl
101 mM
Urea
10,3 mM
Creat
69 uM
eGFR by MDRD
>60 ml/min/m2
eGFR by CKDEPI
97 ml/min/m2
Ca
2,26 mmol/L
Mg
1,03 mmol/L
Phos
1,01 mmol/L
Total prot
73 g/L
Alb
37 g/L
Total bili
3 umol/L
Conj bili
2 umol/L
ALT
15 U/L (10-40)
AST
19 U/L (15-40)
ALP
284 U/L (53 – 128)
GGT
76 U/L (<68)
LD
345 U/L
CRP
52 mg/L (<10)
Total PSA
846.5 ug/L (<4)
TSH
1,33 mIU/L (0.27 – 4.2)
Hb
5.6 g/dL
MCV
88.3 fL
WCC
7.57 cells/uL
Table 1 – Blood results on 06/07/2020
Other Investigations
Chest X-Ray: Left hilar opacities
X-ray of the limbs: Global lytic lesions involving both proximal femurs
Figure 1 – Lytic lesion seen in the centre of the thoracic vertebral body.Figure 2 – Included for comparison with Figure 1 – not as big lytic lesion seen.Figure 3 – MRI image of the same thoracic vertebral body as shown in Figure 1.Figure 4 – Transverse and coronal views of the CT scan with the outline of the prostate marked in yellow (left middle) and purple lines (right top and bottom)Figure 5 – Small lytic lesions visible in the proximal femur.
Prostate biopsy
MACROSCOPY: Specimen consists of two cores, the longest measuring 12mm in length.
MICROSCOPY Sections show 2 prostatic cores, both infiltrated by a prostatic adenocarcinoma.
Metastatic Prostate Carcinoma with multiple metastases to the bones (thoracic spine and both femurs).
Take Home Message
Prostate-specific antigen (PSA, also known as kallikrein III, seminin, semenogelase, γ-seminoprotein and P-30 antigen) is a 34-kD glycoprotein produced almost exclusively by the prostate gland. It is a serine protease enzyme.
Most PSA in the blood is bound to serum proteins. A small amount is not protein-bound and is called ‘free PSA’. In men with prostate cancer, the ratio of free (unbound) PSA to total PSA is decreased. The risk of cancer increases if the free to total ratio is less than 25%.
The lower the ratio is, the greater the probability of prostate cancer. Measuring the ratio of free to total PSA appears to be particularly promising for eliminating unnecessary biopsies in men with PSA levels between 4 and 10 mg/L.
ALP (alkaline phosphatase) is well known to be a marker of ductal hepatic damage. ALP, being an isozyme, however has its origin from various tissue sources in the body. It is present in the liver, bile duct, kidney, bone, intestinal mucosa and placenta. The majority of ALP in serum is from either skeletal or liver origin. In adults the major form is from liver and in children the major form is from the skeleton.
Blood levels of alkaline phosphatase increase by two to four times during pregnancy. This is a result of additional alkaline phosphatase produced by the placenta.
If it is unclear why alkaline phosphatase is elevated, isoenzyme studies using electrophoresis can confirm the source of the ALP. It would likely in this patient be quite clear that the raised ALP would be due to the excess leakage from the osteolitic lesions from the metastases, but who knows, the patient may have had a beer or five in the preceding 3 weeks leading up to the bloods being drawn. The fact that the other liver enzymes are near-normal, makes alcohol consumption less likely though.
A case of severe hypoalbuminaemia
HOSP #
Lab no: SA03948371
WARD
Paediatric Ward
CONSULTANT
Dr. Jody Rusch
DOB/AGE
16 y Female
Abnormal Result
Albumin of 8 g/L
Presenting Complaint
Signs and symptoms of a urinary tract infection made the patient present to a general practitioner.
History
No known chronic medical illness were present upon initial presentation.
No medical treatment was being taken for chronic illnesses.
The patient had reported taking NSAIDS before for pain in the lower abdomen. The exact drug / dose was unknown.
Examination
All clinical findings are unfortunately not available for this patient.
It is known that the patient had been having lower abdominal pains upon presentation (which was not due to pregnancy).
A urinary tract infection was suspected by the initial treating physician. Upon the other finding of edema, investigation towards the cause was investigated.
Typical findings of nephritic syndrome are:
Fever
Edema (due to hypoproteinemia)
High blood pressure (due to activation of the renin-angiotensis-aldosterone system).
Joint pain
Muscle pain
Malar rash
Foamy urine (proteinuria)
Laboratory Investigations
Albumin 14 g/L
Cholesterol 8.14 mmol/L
Urine Protein:Creatinine ratio: 1.62 g/mmol creat
C3: 0.29 (Low)
C4: 0.07 (Low)
Creatinine 255 – 322 umol/L
Other Investigations
Final Diagnosis
Lupus Nephritis with hypoalbuminemia
Take Home Message
The clinical presentation of this patient is a good example of the findings in patients who initially present with renal failure. The extent of renal failure is often so severe, that when the patient presents with signs and symptoms of renal failure, there are quite significant permanent renal damage already.
Patients with nephrotic syndrome present with significant proteinuria with resultant hypoproteinemia, firstly hypoalbuminemia, followed by the other bigger proteins like gammaglobulins, alpha-1, beta-1 and beta-2 (complement) proteins. Because alpha-2 (macroglobulin) comprises one of the biggest proteins (in molecular size) in the serum, it generally stays in the serum relatively longer than the other leaking proteins.
Because the liver increases its production of proteins to try compensate for the reduction in osmolality, the production of VLDL rises significantly and hence Triglycerides (and cholesterol) rises. Thus cholesterol in this patient measured 8.14 mmol/L.
The pathophysiology of lupus nephritis is that of autoimmunity. Autoantibodies direct themselves against nuclear elements. The characteristics of nephritogenic autoantibodies are antigen specificity directed at the nucleosome. High affinity autoantibodies form intravascular immune complexes, and autoantibodies of certain isotypes activate complement. Hence the C3 and C4 which are low often indicates active lupus disease.
Section 7.7 – Laboratory Management
Presentations attended related to laboratory management:
PathCape 2018
Accreditation, Quality and Leadership – Prof A Zemlin – PathCape 17 August 2018
Error Free Lab Work: Is it an achievable target? – Prof Yenice (Turkey) – PathCape 17 August 2018
P4P (pay for performance) in Laboratory Medicine – Dr Orth (Germany) – PathCape 17 August 2018
Laboratory management course – University of Stellenbosch 02-05 November 2020
The laboratory management course which I attended through Stellenbosch University was an exciting experience. Although during COVID times, it wasn’t necessarily a trip to Stellenbosch where one could have a glas of wine at a local wine farm afterwards, but nonetheless it was an amazing experience. I have made friends with colleagues in other Pathology disciplines remotely and we needed to prepare a Strategic Business Plan and present it at the end of the course.
All talks attended were focussed on laboratory management. The skills learnt during this course will likely still bring much joy and productivity into my work life in future and the tools learnt to properly manage a laboratory are of enormous value.
There were tasks from as simple as a left-right quizz, to a QC workshop for our chemical pathology registrars with Levy-Jennings chart interpretation and the lot. Some of the most enjoyable topics for me were: Adding value to lab medicine, a topic often focussed on by Prof Annie Zemlin, effective laboratory leadership, focussed on by Prof Rajiv Erasmus, an effective laboratory leader in Chemical Pathology and the topic on Risk management by Prof Preiser. Together these topics which were presented (see below) made up an astounding course which brought together a few aspects rarely covered by other lecturers or even reading material elsewhere. This is what makes this course a must for future laboratory leaders.
Day 1 – 02 November 2020 Talk 1 – Leadership Skills For Effective Laboratory Management – Mandela’s Lessons – Prof RT Erasmus Talk 2 – Laboratory Organization – Best Practice – Dr Z Chapanduka Talk 3 – Ethical Leadership – Prof RT Erasmus Talk 4 – Strategy And Leadership Strategic And Goal Planning For Effective Laboratory Management – Prof RT Erasmus Talk 5 – Budget And Introduction Of New Tests – Prof AE Zemlin Talk 6 – Laboratory Safety – Prof TS Pillay
Day2 – 03 November 2020 Talk 1 – Leading And Managing Change In The Laboratory – Prof G van Zyl Talk 2 – Leadership And Diversity – Prof AE Zemlin Talk 3 – Preventing And Managing Conflict In The Laboratory – Prof Schneider Talk 4 – Risk Management In The Diagnostic Laboratory – Prof W Preiser Talk 5 – POPIA For The Healthcare Professional – Dr C Swanepoel Talk 6 – EBLM and Audit – Dr T Jalavu
Day3 – 04 November 2020 Talk 1 – Use Of Quality Management Tools To Assess And Improve Quality – Prof A Whitelaw Talk 2 – Iso 15189 And Preparing The Lab For Accreditation – Prof AE Zemlin Talk 3 – Non-Conformances & Document Control – Dr AA Khine Talk 4.1 – Lean Management And Quality – Dr AA Khine Talk 4.2 – Six Sigma Approach To Quality – Dr AA Khine Talk 5 – Managing A POCT Service – Prof A Whitelaw Talk 6 – Method Validation – Dr M Hoffmann Talk 7 – Extra-Analytical Errors – Dr E Kruger
Day4 – 05 November 2020 Talk 1 – Harmonization, Standardization And Traceability – Dr T Jalavu Talk 2 – Electronic Gatekeeping – Dr H Vreede Talk 3 – Medico-legal aspects of laboratory practice and maintenance of the chain of evidence – Prof J Dempers Talk 4 – Autoverification – Dr H Vreede Talk 5 – Demand Management – Prof AE Zemlin Talk 6 – Uncertainty Of Measurement – Dr E Kruger
Checklist for portfolio assessment
Checklist for Portfolio Assessor
Is the portfolio well-organised and indexed? Does it correspond with all the headings and sections?
Has a research protocol been included?
What research projects has the candidate been involved in? Are these of the expected level and scope for the discipline?
Has the candidate provided evidence of journal club presentations and attendance with adequate reflective learning comments appended?
Laboratory training: has the candidate provided evidence of training in laboratory techniques and methods with evidence of reflective learning a. Has the candidate provided a written technical report of methods in the laboratory? b. Do these reports reflect a good understanding of laboratory techniques?
Management training: a. Is there evidence of management training by attendance at seminars, workshops and courses with accompanying reflective comments.
Case studies: Has the candidate complied with the requirements for short and long case reports? Has the candidate provided the required number of short and long cases with indepth analysis for each? Has the candidate provided the following for the long cases (3000-3500 words): Understanding the theory of the case Clinical assessment of the case Proposed additional investigations or comment on appropriateness of the investigations performed Laboratory issues that could influence the work-up Overall summary of the case Presentation in the portfolio, including a literature review with references in a form appropriate to a journal article.
Seminars and lectures delivered by the candidate: has the candidate provided evidence of lectures and seminars by providing copies of presentations?
Has the candidate performed any laboratory audits?
What outputs has the candidate provided (audits, papers, SOPs) At least one audit (3000-3500 words excluding references) 1000-1500 word description of the MMed project if not completed or attach reprint of paper or submitted manuscript
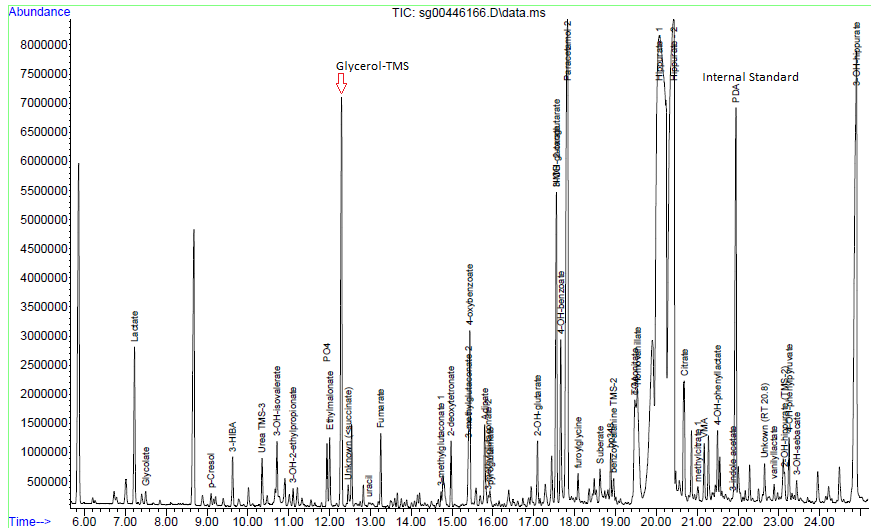
Glycerol which is significantly raised on urine organic acid analysis.
Figure 1 – Chromatogram. The high levels of Glycerol (with TMS – trimethyl silyl derivative) which is >0.5 the peak height of the internal standard (PCA – pentadecanoic acid).Figure 2 – Mass spectrum of the peak as indicated by Glycerol TMS above in Figure 1.Figure 3 – Follow up gas chromatogram without KY-jellyFigure 4 – Mass spectrum in the peak marked as “Glycerol-TMS” from figure 3.
Presenting Complaint
Patient is a 3 month old male with signs and symptoms of sepsis.
History
Patient presented with significant failure to thrive.
Laboratory Investigations
Triglycerides : 4.47 mmol/L
Other Investigations
Faecal elastase 81 ug/g stool
Reference range (adults and children > 1 month):
> 200 ug elastase/g stool: Normal exocrine pancreatic function
100-200 ug elastase/g stool: Moderate/mild pancreatic insufficiency
< 100 ug elastase/g stool: Severe exocrine pancreatic insufficiency
These ranges apply to formed stool samples. Watery stool samples may yield spuriously low elastase results due to dilution, and a formed stool sample should be sent for re-analysis.
Final Diagnosis
Glycerol contamination of the skin – as excluded by the repeat analysis.
Take Home Message
Glycerol (glycerine) is a common contaminant of urine organic acids due to being present in various skin products / creams. Contamination can be eliminated by thorough cleaning of the perineum with normal saline or doing an “in-out” catheterization procedure for urine collection in neonates. Interestingly glycerol is also one of the main ingredients in KY jelly, a common lubricant use for catheterization.
High glycerol in serum will present with a falsely high triglyceride level on most routine chemistry analysers due to the inherent enzymatic conversion of triglycerides to glycerol before further steps to measurement.
Figure 3 – Explanation of triglyceride determination by amperometric detection.
Sepsis is more common than inherited metabolic diseases and so is pre-analytical caveats such as glycerol contamination of the perineal skin.
Hyperaldosteronism with Hyperreninaemia in a 15 year old
The aldosterone:renin ratio (ARR) is a screening test for primary hyperaldosteronism and is most sensitive when both an absolute aldosterone > 350 pmol/L and an ARR > 118 pmol/ng is present.
Na: 138 mmol/L
K: 4.5 mmol/L
Urea 3.7 mmol/L
Creatinine: 49 umol/L
Total Calcium: 2.55 mmol/L
Urine dipstick 1+ protein
The urine protein:creatinine ratio was 0.044 g/mmol creat
Presenting Complaint
Patient presented with a 2 week history of blurry vision, intermittent headaches, hot flushes and mild intermittent epistaxis.
History
No known comorbidities
Multiple episodes of otitis media as a child
Presented with hypertension and evidence of target organ damage (retinopathy and left ventricular hypertrophy).
Funduscopy: Silver wiring, hard exudates, no haemorrhages, no papiloedema
CVS: bounding, peripherals pulses present. JVP raised, Undisplaced apex. Regular HR, no Radial/ femoral delays. Normal S1 & S2 with flowmurmur grade 2.
GIT: soft, non-tender. Ballotable left kidney, non-tender
Resp: Central Trachea, clear viscular breath sounds, no added sounds.
Neurological Exam: GCS 15/15, normal higher functions, no focal neurological signs.
BP control achieved with Doxazocin (increased to 4mg dly) and Atenolol (increased to 50mg dly)
Laboratory Investigations
TestItem
Value
Units
Reference Range
Urine collection period
24
hours
Urine volume
1280
mL
Urine metanephrine
350
nmol/L
Urine normetanephrine
16350
nmol/L
dU metanephrine
448
nmol/24 hrs
167 – 938
dU normetanephrine
20928 (High)
nmol/24 hrs
311 – 1562
Urine metanephrine : creat ratio
159 (High)
nmol/mmol creat
17 – 88
Urine normetanephrine : creat ratio
7432 (High)
nmol/mmol creat
23 – 176
Table 1 – Urine metanephrines (fractionated)
Other Investigations
ECG: Biatrial enlargement, left ventricular hypertrophy
Chest X-ray: Normal Cardio-thoracic index
Cardiac Ultrasound: Concentric left ventricular hypertrophy with preserved left ventricular ejection fraction. No valvular pathology.
KUB ultrasound: Similar kidney sizes. A mass with a cystic center was noted with no evidence of metastatic disease. Diagnosis suggested to be most likely a pheochromocytoma.
Final Diagnosis
Right-sided phaeochromocytoma
Take Home Message
Aldosterone : Renin ratio cannot be looked at alone. A raised value in either of the Aldosterone and Renin should be investigated further, especially if severely deranged like in this case.
Always investigate hypertension in a child until the cause is found. Hypertension in a child is not normal.
3 consecutive 24 hour urine collection samples are recommended for diagnosis of phaeochromocytoma as some tumours only secrete epinephrines / norepinephrines episodically. In this case however it was not necessary as the case was clear with a markedly raised dU-normetanephrine level.
Screening for pheochromocytoma is an essential part of the workup for secondary hypertension. Urinary vanillyl mandelic acid (VMA) was traditionally used to diagnose phaeochromocytoma. It has a low sensitivity (60-70%). Later, catecholamines measurement in plasma (PCAT) and urine (UCAT) emerged as useful tests. The sensitivity of catecholamines is limited by their episodic release from the tumour cells. The sensitivity ranges from 76-86 % for PCAT and UCAT and the specificity is around 81-99 %.
Metanephrines are methylated metabolites of catecholamines.
Metanephrines are secreted continuously from the tumour cells, independent of the intermittent release of catecholamines. The metanephrines are metabolized by conjugation, primarily in the hepatomesentric organs. Plasma metanephrines (pMN) are measured in the free form (not routinely offered in South Africa) whereas urinary metanephrines (uMN) represent mainly the conjugated form. Hence compared to pMN, uMN is less specific. Studies have shown that plasma free metanephrines have a sensitivity of 96-100 per cent and specificity of 85-100 % superior to that of uMN which has a sensitivity of 93-99.6 per cent and specificity of 71-77 per cent.
Previous methods used colorimetry or spectrophotometry as total MET (metanephrine + normetanephrines) which includes a combined measurement of metanephrine (MN) and normetanephrine (NMN). These methods were superseded by liquid chromatographic assays (LC) that allow individual measurement of MN and NMN.
At Red Cross Hospital Laboratory we use a gas chromatography with mass spectrometry, which is not so widely used for fractionated metanephrines. An isotope dilution method is employed, bringing the method up to internationally recognized standards and the quality assurance of the method at the Red Cross Chemistry lab performs well on the international EQA scheme used.
A case of gas
HOSP #
WARD
CONSULTANT
John Stanfliet / Heleen Vreede
DOB/AGE
52 y Female
Abnormal Result
Oral lactose tolerance test:
Time
Fasting
15’
30’
45’
60’
90’
Blood glucose (mmol/L)
4.3
4.3
4.2
4.5
4.4
4.6
Presenting Complaint
Patient presented with bloating and abdominal pains.
History
The medical history is not known
Patient reported symptoms of bloating and abdominal pains. Upon further questioning it became known that she had been troubles by these symptoms especially severe after consuming dairy products.
Examination
Not known.
Signs and symptoms often associated with this condition are:
Abdominal pain and bloating
Gass / flatulence
Diarrhoea
Constipation
Laboratory Investigations
Time
Fasting
15’
30’
45’
60’
90’
Blood glucose (mmol/L)
4.3
4.3
4.2
4.5
4.4
4.6
Other Investigations
No other investigations were done.
Final Diagnosis
Patient appears to be lactose intolerant since we expect a rise of >1.7 mmol/L in glucose when lactose is adequately digested and absorbed after a lactose load of 50g.
The following cut-offs are frequently used:
Glucose rise > 1.7 mmol/L at any of the time points is a normal lactase response
Glucose rise of 1.1-1.7 is equivocal
Glucose increases < 1.1 is consistent with lactase deficiency
Take Home Message
Lactose consists of Glucose and Galactose.
The biochemical handling of oral lactose
The disaccharide cannot be absorbed and needs to be cleaved before absorbtion by the enterocyte. This happens by lactase, normaly present on the distal part of the brush border of the enterocyte. These enterocytes can become damaged by enteritis and loose activity of lactase partially. Lactase activity also decreases with age, hence elderly do develop partial lactose intolerance. The transporter that carries glucose and galactose into the enterocyte is the sodium-dependent hexose transporter, SGLT-1. As the name indicates, this molecule transports both glucose and sodium ion into the cell and in fact, will not transport either alone.
SGLT1 shouldn’t be confused with a GLUT.
Inhibition of SGLT1 delays and reduces glucose absorption in the small intestine, thus improving post meal glycemic control. This is beneficial particularly in patients with declining renal function where SGLT2 inhibition is less effective.
SGLT2 is the major transport protein and promotes reabsorption of glucose from the glomerular filtration back into circulation and is responsible for approximately 90% of the kidney’s glucose reabsorption.
SGLT2 inhibitors, also called gliflozins, are a class of medications that alter essential physiology of the nephron; unlike SGLT1 inhibitors that modulate Sodium/Glucose channels in the intestinal mucosa.
Figure 1 – Picture illustrating a hexose binding to an SGLT protein
How does lactose intolerance normally present?
Bloating and abdominal cramps after dairy ingestion. (Bacteria metabolise disaccharides and produce H2, hence don’t light your farts when lactose intolerant.) It is less pronounced with dairy which has a lot of lactobacilli (live cultures) in it since they can partially digest some of the lactose. Thus cheese and joghurt doesn’t give such severe Sx as the raw milk products.Good. For your own education, think about sour milk
What is the difference between lactose intolerance, milk allergy and galactosaemia?
Lactose intolerance is explained above, and can be either primary (defective enzyme) or secondary due to another condition as is the case in enteritis where the enterocytes are regenerating and temporarily expressing less enzyme on the apical brush border.
Milk allergy is an abnormal response by the immune system to milk and products containing milk. It’s one of the most common food allergies in children. Cow’s milk is the usual cause of milk allergy, but milk from sheep, goats, buffalo and other mammals also can cause a reaction. The allergy is most likely to one of the exogenous proteins in animal origin milk. Milk allergy presents like any other allergy with hives and/or urticaria.
Galactosemia is the inability to metabolise galactose to glucose for metabolism.
3 genes can contain a mutation (GALT, GALK1, and GALE) coding for:
Galactose-1-P-uridylTransferase
GalactoKinase
UDPGalactose Epimerase
A case of crystals in the cornea, but wait first…
HOSP #
WARD
Red Cross Padiatric Hospital
CONSULTANT
George van der Watt / Surita Meldau
DOB/AGE
1y 5m
Abnormal Result
Inorganic phosphate 0.85 L mmol/L (1.00 – 1.95)
Presenting Complaint
It is unknown what this patient’s presenting complaint was.
Common complaints in similar patients are:
Photophobia (see slit lamp picture below, which explains why).
Bone pain
Weakness
Slit lamp view: Crystals in the cornea – this is typical of patients with cystinosis. Cystine crystals deposit inside the corneal cells as they cannot adequately export Cystine out of their lysosomes.
History
The items below illustrates the recorded information on the laboratory request forms as they were captured by our lab staff on the respective samples:
SEPSIS
Vit D Deficiency
Vit D Deficiency
HYPOCALCAEMIA
?LOSSES
?PTB
?PTB
?FANCONI
?SEPSIS
?SEPSIS
?SEPSIS
? FANCONI SYNDROME
? Cystinosis
Risk factor:
Contact
Cough
Fanconi's ? cystinosis
Hypocalcaemia
Pneumonia.
Pneumonia.
Fanconi syndrome
? Hypoglycaemia
post iv calcium for hypo
Sepsis. Cystinosis
Sepsis. Cystinosis
Cystinosis
FANCONIS SYNDROME
Fanconi's syndrome
Cystinosis
Cystinosis
Sepsis. Fanconi syndrome, hypocalcaemia
Hypocalceamia
CYSTINOSIS- HUNGRY BONE D
It can be seen above that the clinicians were noting Fanconi’s syndrome and were querying Cystinosis.
Examination
Data not available. Generally patients with Fanconi’s syndrome present with loss of electrolytes from the proximal and distal renal tubuli:
Nephropathic cystinosis in untreated children is characterized by renal Fanconi syndrome, poor growth, hypophosphatemic/calcipenic rickets, impaired glomerular function resulting in complete glomerular failure, and accumulation of cystine in almost all cells, leading to cellular dysfunction with tissue and organ impairment. The typical untreated child has short stature, rickets, and photophobia. Failure to thrive is generally noticed after approximately age six months; signs of renal tubular Fanconi syndrome (polyuria, polydipsia, dehydration, and acidosis) appear as early as age six months; corneal crystals can be present before age one year and are always present after age 16 months. Prior to the use of renal transplantation and cystine-depleting therapy, the life span in nephropathic cystinosis was no longer than ten years. With these interventions, affected individuals can survive at least into the mid-forties or fifties with satisfactory quality of life.
Intermediate cystinosis is characterized by all the typical manifestations of nephropathic cystinosis, but onset is at a later age. Renal glomerular failure occurs in all untreated affected individuals, usually between ages 15 and 25 years.
The non-nephropathic (ocular) form of cystinosis is characterized clinically only by photophobia resulting from corneal cystine crystal accumulation.
Because the thyroid glands are actively translating and resorbing thyroglobulin, the thyrocytes (thyroid colloid epithelial cells) are prone to accumulation of cysteine within their lysosomes, hence these children often also develop hypothyroidism with a palpable thyroid gland.
Laboratory Investigations
Biochemistry:
As can be seen on these set of results, the calcium persistently measured low with initial presentation. Newest results are to the left, oldest on the right.Later it can be seen that significant hypophosphatemia also developed.
Genetic screening
PCR with enzyme digest:
Enzyme digest – Expected fragments The PCR product before restriction enzyme digest is 261 bp long. Without the mutation, the restriction enzyme will cut this product in 2 places, yielding 3 fragments 179, 42 and 40 bp. (Normal) With the mutation, the restriction enzyme will cut the PCR product in 3 places, yielding 4 fragments, 135, 44, 42 and 40bp.
1st PCR with enzyme digest:
L – Ladder (100 bp at bottom with increments of 100 bp to the top) 1. Patient sample 2. Positive control 4. Normal (Negative) control 5. Blank
Upon the first PCR for a gene screen which was done, there occurred some evaporation in the third tube (lane no. 3 – positive control), upon incubation with the enzyme digest mix. This incubation step is minimum 3 hours at 37 degrees Celcius and do not have the high temperatures associated with the PCR process. Thus the reason for evaporation remains unclear and may have been an incompletely closed lid or a defective PCR tube rim or cap.
It is not expected to see an undigested PCR product (>200bp) as can be seen in the top band in lane 3, hence it is clear that there was incomplete digestion of PCR product in this PCR tube. Even though it is clear that the patient sample ( lane 1) represents a homozygous positive result (lane 2), one cannot authorize results when both the positive and normal control worked well.
Thus the PCR was repeated.
2nd PCR with enzyme digest:
L – Ladder (100 bp at bottom with increments of 100 bp to the top) 1. Patient sample 2. Positive control 3. Positive control with 2x enzyme mix added 4. Normal control 5. Blank Image intentionally left uncropped and long vertically: Note the cloudy grey portion below the bottom most bands **see “Take Home Messages” below
In the second PCR it can be well seen how the Positive control did undergo complete digestion. No evaporation was noted in this run.
Other Investigations
Leucocyte cystine is another investigation which helps make the diagnosis in patients with presumed cystinosis.
Leucocyte cystine report in this patient:
Protein 0.58 g/L
Leucocyte Cystine 1.16 nmol/mg protein
Reference range:
Normal < 0.1 nmol cystine/mg protein
Cystinosis > 1.0 nmol cystine/mg protein
Cystinosis on Rx (target level) < 0.5 nmol cystine/mg protein
Our reporting comment on the Lab Information Systems reads as follows:
Please note that the diagnosis of cystinosis can be confirmed in the majority of South African patients by screening for the common South African Black mutation CTNS-c.971-12G>A which results in an estimated newborn incidence of 1/10 000 in this population. A molecular diagnosis is of value in that siblings of index cases can be screened and identified for early intervention which improves the outcome in this disorder.
Final Diagnosis
Patient is homozygous positive for cystinosis by the common South African mutation, as confirmed on leucocyte cystine as well as on the gene screen.
Take Home Message
** The grey portion at the bottom of the electropherogram – this indicates the movement of the ethidium bromide out of the gel towards the cathode. One should ideally not let a gel run beyond the intended time period as the migration of ethidium bromide “dye front” beyond the smallest band may cause band to “de-stain” and not be visualized well.
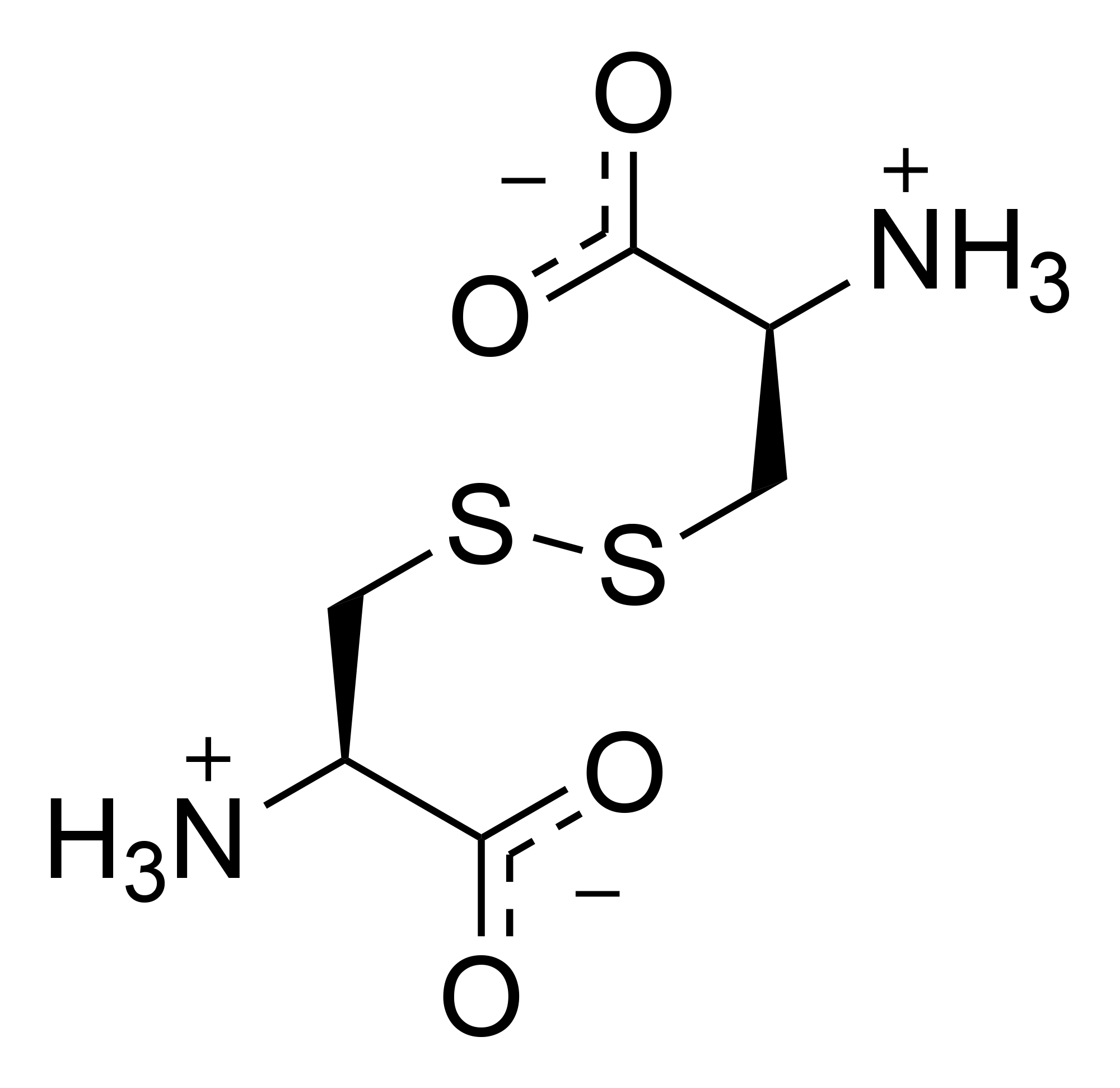
The CTNS gene provides instructions for making a protein called cystinosin. This protein is located in the membrane of lysosomes. Proteins digested inside lysosomes are broken down to amino acids. These are then moved out of lysosomes by transport proteins. Cystinosin is a transport protein that specifically moves the amino acid in its dimeric form cystine out of the lysosome.
Cystine is a sulfur-containing amino acid obtained by the oxidation of two cysteine molecules which are then linked via a disulfide bond.
More than 80 different mutations that are responsible for causing cystinosis have been identified in the CTNS gene. The most common mutation is a deletion of a large part of the CTNS gene (sometimes referred to as the 57-kb deletion), resulting in the complete loss of cystinosin. This deletion is responsible for approximately 50 percent of cystinosis cases in people of European descent. Other mutations result in the production of an abnormally short protein that cannot carry out its normal transport function. Mutations that change very small regions of the CTNS gene may allow the transporter protein to retain some of its usual activity, resulting in a milder form of cystinosis.
The treatment / management of patients with cystinosis includes Cysteamine, a drug which binds cysteine and forms Cysteamine-cysteine (see figure above). This molecule is similar in structure to lysine and can be exported from the lysosomes by a lysine transporter.
Interestingly, South Africa likely has the highest incidence of cystinosis in the world due to a common mutation, G > A mutation in intron 11 of the CTNS gene (c.971-12G > A p.D324AfsX44), likely due to some sort of founder-effect in black and coloured patients: https://link.springer.com/article/10.1007/s00467-014-2980-7 I’m proud of this article as it was published by scientists at our institution.
A useful web site to learn nomenclature of gene variants is HGVS.
Storage disease on Autopsy
HOSP #
WARD
Histopathology
CONSULTANT
Dr. Jody Rusch
DOB/AGE
1 week Female neonate
Abnormal Result
An email from a colleague and mentor summarizes the abnormal result the best:
I am not sure if this will be an easy one to nail down without extensive testing or luck.
To summarise:
Female neonate No mention of ethnicity C-section – hydropic on US Birth weight 2935 g
Low Apgars (2 and 5) Intubated, ventilated, ICU Did not grow Demised on day 7 of life in ICU
Non-immune heart failure plus storage disorder: IMDs can cause heart failure. Most likely lysosomal storage disease (14 different ones have been associated with HF) Most LSDs are AR inheritance HF, facial dysmorphism, AR inheritance, previous sibling hx Common in European populations (and it appears globally) include – Mucopolysaccharidosis type VII, Gaucher’s disease, and GM1-gangliosidosis In SA: Gaucher’s disease (Ashkenazi-Jewish population) – GD2 should be considered in severe perinatal with HF
Extensive list of Lysosomal storage diseases associated with heart failure: Gaucher disease, type II, Morquio disease, Hurler syndrome, Sly syndrome, Farber disease, GM1 gangliosidosis, I-cell disease, Niemann-Pick disease type A and type C, Infantile Sialic Acid Storage disorder, alpha-neuroaminidase deficiency, multiple sulfatase deficiency, and Wolman disease.
Consider also non-lysosomal diseases Other IMDs: Type IV (Anderson disease) Congenital disorders of glycosylation Zellweger syndrome LCHAD Primary carnitine defic Smith Lemli Opitz Syndrome
Also hypothyroidism
If a specific diagnosis (beyond likely LSD) is required, and will be paid for, perhaps Invitae have a panel? Hopefully Prof can weigh in on this and help guide further testing.
Kind regards J
Presenting Complaint
The histopathologist contacted me regarding any “screening tests” for lysosomal storage diseases
History
Maternal hx: 39 yr old Booked – normal bloods 35 weeks gestation Previous pregnancy – stillbirth due to hydrops foetalis (normal karyotype) No mention of consanguinity
Examination
Post mortem: Eyes wide set Left Ear malformed Flattened nasal bridge Hydrops foetalis (HF) Steatosis – lung, liver, heart, placenta
Laboratory Investigations
Not available
Other Investigations
Not available
Final Diagnosis
Unknown
Take Home Message
Message from Prof David Marais:
Hi S & J Interesting and I wish we could devote much more effort to solve these cases. Especially since this is the second time this mother has had this sad experience and the next pregnancy may result in the same.
On first principles:
This appears autosomal recessive
The dysmorphology eliminates many “simpler” inherited errors as homeostasis through the placenta settles imbalances. However, errors involving tissue differentiation, structural components may have dysmorphology. E.g. sterol synthetic defects, mucopolysaccharidoses…It is easy to exclude Smith Lemli Opitz with 1mL serum or plasma even at this stage. However, syndactyly is a very strong feature and hydrops is uncommon but described. Happy to do this if sample is available. Mt abn has been described as well and might explain steatosis though not likely.
Microvesicular steatosis in several organs is suggestive of incomplete mobilisation of FA into mitochondria for oxidation or inadequate oxidation in mitochondria. These disorders do not usually result in dysmorphology and it is said renal steatosis is typical. LCHAD deficiency has caused hydrops but not dysmorphism to the best of my knowledge. Wolman’s disease has adrenal calcification but not hypoplasia as far as I know and not typically hydrops and diffuse steatosis – will need to check this again. The steatosis could be secondary to severe metabolic stress.
Hydrops fetalis should be taken as a strong clue. The lysosomal disorders can cause these. The list I found in JIMD Reports (2018) Hurler syndrome (MPS-I; OMIM #607014), Morquio-A (MPSIVA; OMIM #253000), Sly syndrome (MPS-VII; OMIM #253220), galactosialidosis (OMIM #256540), sialidosis (OMIM #256550), GM1 gangliosidosis (OMIM #230500), Gaucher type 2 (OMIM #230900), Niemann-Pick disease types A and C (NPD-A and NPC; OMIM #257200, 257220), Farber granulomatosis (OMIM #228000), Wolman disease (OMIM #278000), mucolipidosis II (I-cell disease; OMIM #252500), sialic acid storage disease (ISSD; OMIM #269920), and multiple sulfatase deficiency (OMIM #272200) which have been shown to be associated.
Interesting that the spleen is absent and that the adrenals are small. This is hard to explain on any of the metabolic disorders above but I shall have to read more extensively.
It may be worth testing the urine or other fluids for sialic acid. Infantile Salla disease is a possibility. (Sialin defect, coarse facies, hydrops fetalis, vacuolated lymphocytes but I was not aware of steatosis.) I can look up if this is practicable with the old fashioned assays and chemicals that we do have. I know I tried to analyse sialic acid in the early 1990s. Will look this up too.
One hopes that the anat path dept keeps samples for work-up. Very important to involve the chem path early if any metabolic disease is suspected as one can do fibroblast biopsy up to a few days in the morgue.
Regards D
Professor Emeritus AD Marais Chemical Pathology 6.33 Falmouth Building University of Cape Town Health Sciences Anzio Rd, Observatory, 7925 Cape Town, South Africa
A case of hypertriglyceridemia with Diabetes mellitus
HOSP #
WARD
Albow Gardens Clinic
CONSULTANT
Prof. David Marais
DOB/AGE
31 y Male
Abnormal Result
31 y/o Male
Presenting Complaint
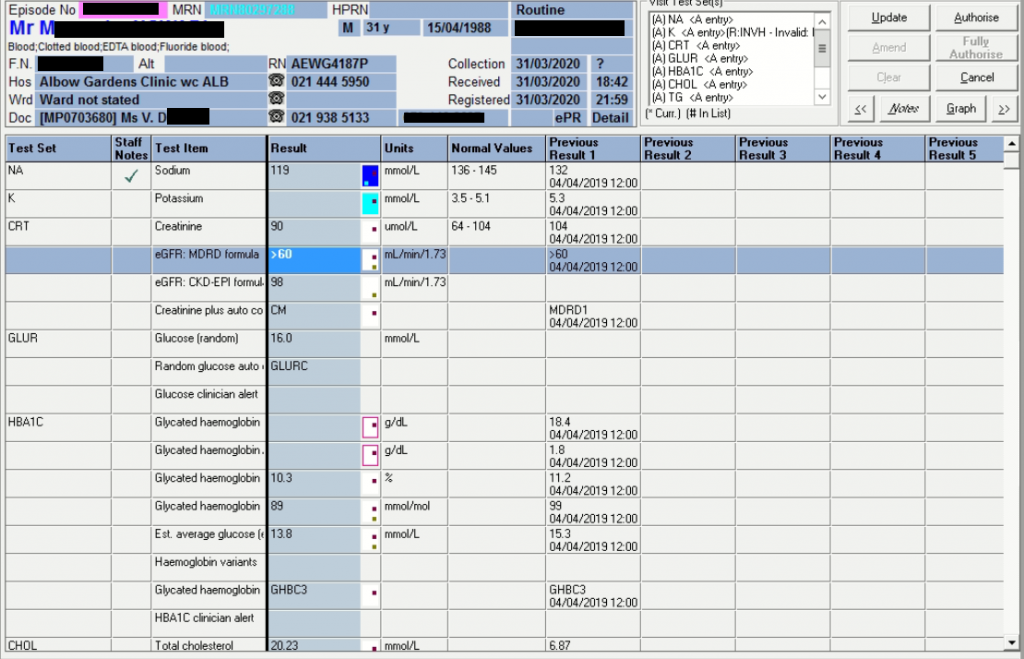
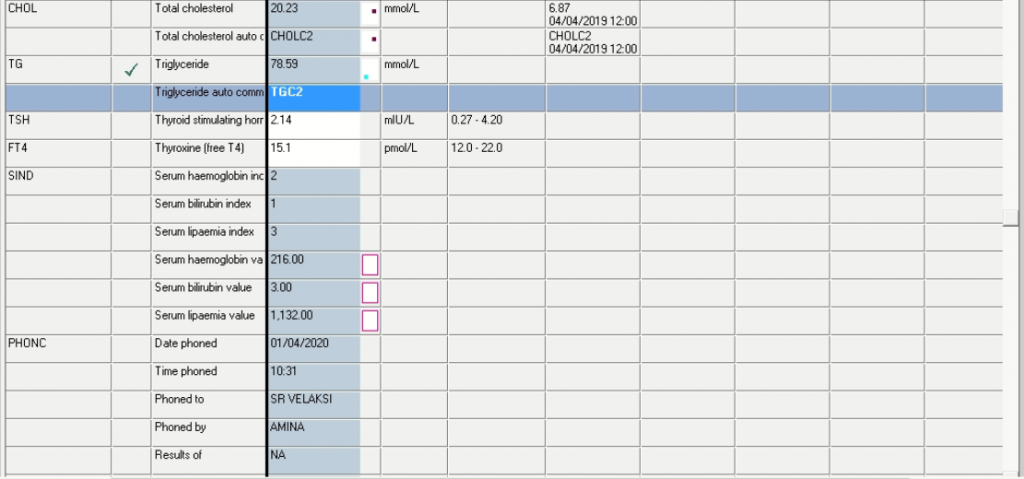
Triglycerides of 78.59 mmol/L
Lipaemia index 3 (value of 1132)
It is likely that the results as set out above was due to a routine follow-up, but unfortunately little clinical information was given by the clinician.
History
The patient is hypertensive and diabetic on treatment since 2018. No other clinical information was given and the drug list was not supplied.
Examination
N/A – No signs and symptoms obtained.
Laboratory Investigations
Other Investigations
We would have loved to do lipid electrophoresis and see better investigations into the cause of the diabetes, but at the time of writing, 14/05/2020, the patient has unfortunately not had the opportunity to follow-up and it can unfortunately not be shown.
In an adult diabetic one would however expect the lipid electrophoresis to be that of a Fredrickson type V.
Hyperlipoproteinemia type V, also known as mixed hyperlipoproteinemia, familial or mixed hyperlipidemia, is very similar to type I, but with high VLDL in addition to chylomicrons.
It is also associated with glucose intolerance and hyperuricemia.
Final Diagnosis
Considering most factors known, and as explained via feedback from Prof. Marais below, diabetes is likely type 2 related to insulin resistance. One should also consider metabolic errors such as glucokinase deficiency causing MODY. Other causes, but unlikely, are endocrine pancreatic insufficiency which could include mitochondrial defects or herbicide-induced diabetes or (post-traumatic) excision of tail of pancreas. HbA1c shows prolonged exposure to high glucose concentrations: 10.3% and 11.2% the year before.
Take Home Message
The following were my thoughts on causality of the high triglycerides initially:
Increased intake:
Overeating (unlikely for this high Triglyceride level), that is why one has lipoproteins – to keep the fat in the blood low and store the fat in liver and tissues.
Excess alcohol consumption, but I’m also not sure (wasn’t sure) if excess alcohol will raise triglycerides this high – it likely may (after prof’s email I think this is very likely the main cause in this patient).
Increased production:
Kidney failure (nephrotic syndrome) (Prot: Creat ratio will likely exclude this – if borderline, a protein electrophoresis can be done).
Decreased metabolism:
Some forms of familial hyperlipidemia such as familial combined hyperlipidemia
Lipid electrophoresis will delineate the Fredericksen Class.
Familial combined hyperlipidemia:
Lipid electrophoresis will show lipoproteinemia type IIB.
LPL deficiency:
Lab tests show massive accumulation of chylomicrons in the plasma and corresponding severe hypertriglyceridemia. Typically, the plasma in a fasting blood sample appears creamy (plasma lactescence).
The absence of secondary causes of severe hypertriglyceridemia (like e.g. diabetes, alcohol, estrogen-, glucocorticoid-, antidepressant- or isotretinoin-therapy, certain antihypertensive agents, and paraproteinemic disorders) increases the possibility of LPL deficiency. Also other loss-of-function mutations in genes that regulate catabolism of triglyceride-rich lipoproteins (like e.g. ApoC2, ApoA5, LMF-1, GPIHBP-1 and GPD1) should also be considered. (remember our case – I won’t mention her name though, patient’s name begins with a K… and ends with …ana).
The diagnosis of familial LPL deficiency is finally confirmed by detection of either homozygous or compound heterozygous pathogenic gene variants in LPL with either low or absent lipoprotein lipase enzyme activity (Jody and I have done this assay with Bharati and Prof once for above patient).
Lysosomal acid lipase deficiency (LAL-D) (aka cholesteryl ester storage disease) – Unlikely – would rather present earlier – the accumulation of fat in the walls of the gut in early onset disease leads to serious digestive problems including malabsorption, the gut fails to absorb nutrients and calories from food. Because of these digestive complications, affected infants usually fail to grow and presents with failure to thrive. As the disease progresses, it can cause life-threatening liver dysfunction or liver failure). Until 2015, apparently there was no treatment (not sure if this is true though), and very few infants with LAL-D survived beyond the first year of life.
I think the clinical presentation and examination and history is much needed before any further investigations are advised.
Also, one should appreciate the size difference which is partly responsible for the electrophoretic mobility of lipoproteins on a gel.
Lipoprotein size illustration
Feedback from Prof. David Marais:
Hi Dieter
Thanks for distributing the interesting case information. The patient is at very high risk of developing acute pancreatitis. Hopefully the medical officer will be able to get in touch with the patient and urgently:
(1) control diabetes mellitus and
(2) restrict dietary fat intake to 10g/d for a few days whereafter 30-40g/d.
(3) restrict alcohol intake to preferably zero or certainly <20g/d.
(4) prescribe fibrate.
(5) Referral to the lipid clinic – unfortunately may take time owing to shut-down of out-patients clinics in the precautions against corona virus spread.
Such severe hyperTGaemia seen in the neonate, infant or child is most likely due to an error in the lipolytic system and all of these are recessively inherited. LPL deficiency is the commonest but there may also be apoCii, apoAv, GPIHBP1 or LMF1 deficiency. In adolescent and young adults the same causes apply but also auto-immune LPL inhibition. In these cases all the agarose gel electrophoresis for lipoprotein separation will display a type I pattern. The highest TG conc I have seen in a patient was 695mmol/L and at 6 weeks of age.
In adults the lipoprotein electrophoresis pattern will usually be a type V. Here, there is usually partial lipase deficiency (often polygenic heterozygotes of LPL system components) and a dietary or metabolic stress. Diet containing triglycerides in large amounts and alcohol. Metabolic stress is mostly diabetes with increased return of NEFA to the liver and export as VLDL. Occasionally, apoE2/2 status with impaired remnant clearance can have a backlogue effect to raise VLDL and chylomicrons. Rarely, in partial lipodystrophies the adipose tissue does not take up NEFA from LPL and the liver puts out more VLDL which competes with chylomicrons for lipolysis. Typically this is associated with diabetes as well. Significant hypothyroidism and renal impairment appear to be excluded as potential secondary causes.
The results indicate long-standing diabetes and hyperlipidaemia. There is likely pseudohyponatraemia. This is because the aqueous part of the aliquot for analysis can be significantly less than the whole volume. The response is to do highspeed or ultracentrifugation so that the lipid can float and the infranatant plasma can be best analysed. Obviously, the whole plasma should be first assayed (in dilution with saline) to be certain of the TG concentration. Alternatively, the lipid volume can be calculated by converting the mmol/L of TG + CE + phospholipid to mass/L and then using the specific gravity of 0.92 g/mL to obtain the volume correction. For practical purposes only the TG and cholesterol values may be used as we do not routinely measure the phosphatidyl choline. Average MW of TG =850da, of CE = 650da, of PL = 750da. Note that usually 70% of cholesterol is esterified. Cholesterol MW = 387da.
Per L, TG of 79mmol/L is 67g, CE of 14mmol/L is 9g, total lipid is 85g.
Each g being 1.09mL, makes this 92.4g/L or 9.24g/100mL. This means that the aqueous portion of the aliquot is about 10% too low. This makes the calculated Na+ concentration about 131mmol/L which is still not normal but certainly closer to the reference range. But the calculation is not highly accurate; partly because PL has not been taken into account; unesterified cholesterol is quantitatively less important.
Diabetes is likely type 2 related to insulin resistance but at this age and especially if dominantly inherited, consider metabolic errors in MODY such as glucokinase deficiency. Unlikely endocrine pancreatic insufficiency which could include mitochondrial defects or herbicide-induced diabetes or (post-traumatic) excision of tail of pancreas. Not certain if patient is on IV line that could provide lipid (Intralipid in parenteral nutrition) or glucose. Regardless, HbAic shows prolonged exposure to high glucose concentrations.