Intellectual disability
| HOSP # | WARD | Endocrine clinic | |
| CONSULTANT | Prof George van der Watt | DOB/AGE | 17 year old boy |
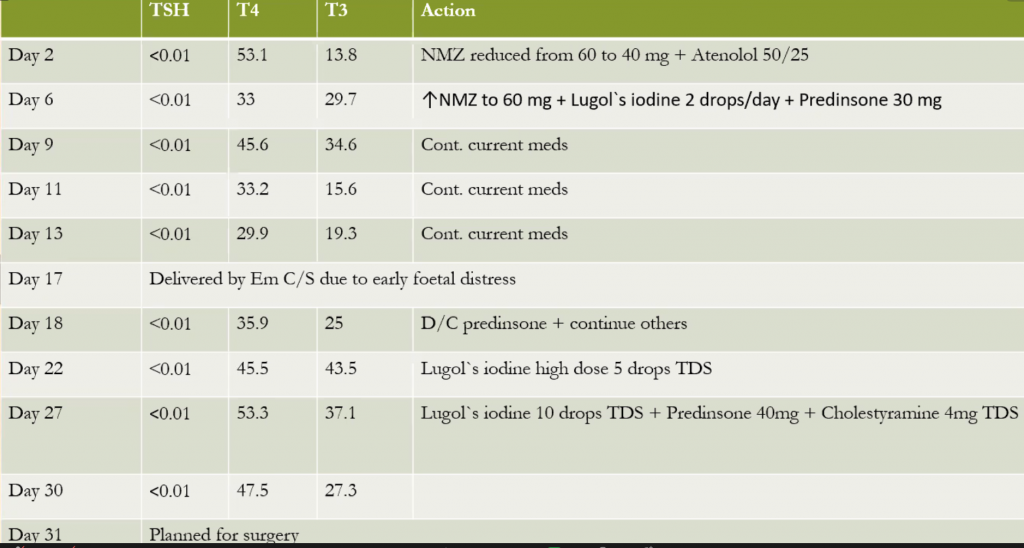
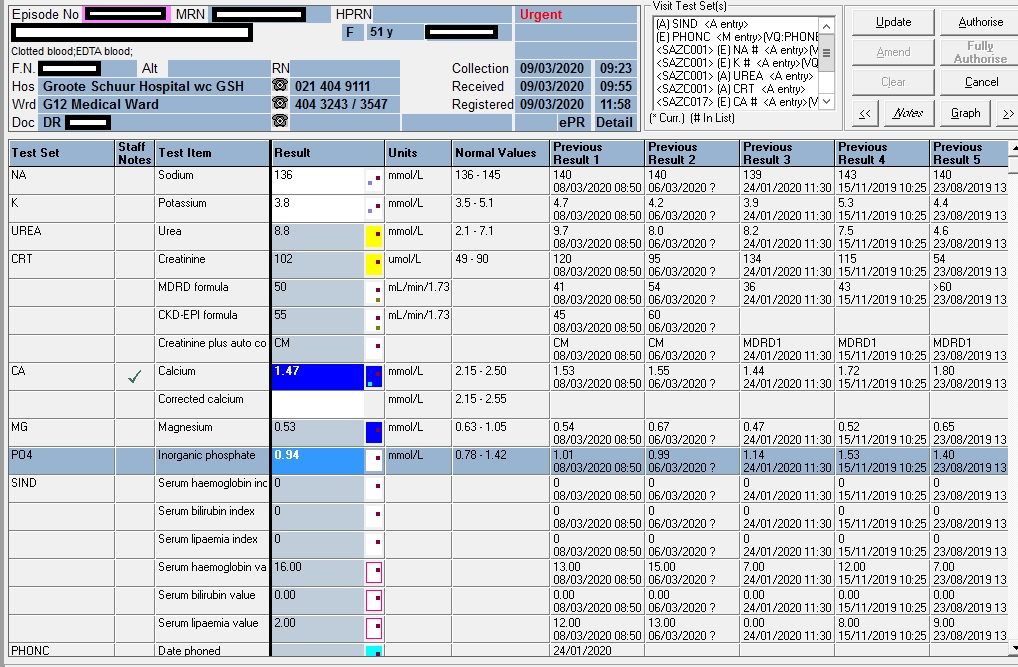
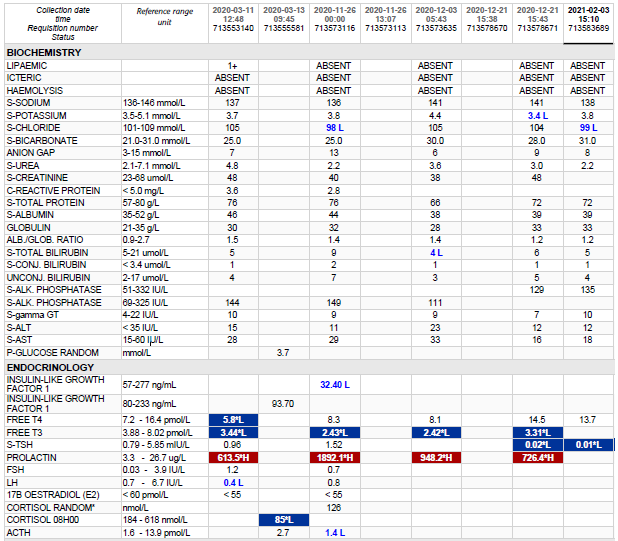
Abnormal Result
High TSH, low T4.
Presenting Complaint
Intellectual impairment
History
This 17 year old boy from Zimbabwe arrived in South Africa at +/- 8 years of age – diagnosed with hypothyroidism (thyroid dysgenesis)
The thyroid functions have never been well-controlled.
Examination
The patient had significant intellectual impairment – impairment in communication, mother struggling to take care of him, and severly impaired social skills.
Laboratory Investigations
Useful tests to perform to assess for the source of thyroid hormone dysgenesis include:
Tests to assess pituitary function:
- ACTH (or cortisol)
- TSH (would generally be low in pituitary causes of cretinism)
- LH
- FSH
- Growth hormone
- Prolactin (posterior pituitary)
Other Investigations
Thyroid scans (scintigraphy) is not required for diagnosis, but may provide important information on the position of the thyroid.
Thyroid ultrasound is an alternative to determine location of the thyroid gland in the neck, but may fail to detect some tissues.
A lateral radiograph of the knee may be obtained to look for the distal femoral epiphysis; this ossification center appears at about 36 weeks’ gestation, and its absence in a term or postterm infant indicates prenatal effects of hypothyroidism.
Final Diagnosis
Thyroid dysgenesis
Take Home Message
Thyroid biological functions
- Control of basal metabolic rate
- Enhancement of mitochondrial metabolism
- Neural development and growth
- Sexual maturation
- Adrenergic stimulation
- Protein synthesis and carbohydrate metabolism
- Synthesis and degradation of cholesterol and triglycerides
- Increases calcium and phosphate metabolism
Congenital Hypothyroidism’s incidence appears to be 1:2000 – 1:4000 live births – Likely the most common preventable cause of intellectual disability worldwide.
Types / causes:
- Thyroid dysgenesis
- TSH Resistance
- Disordered thyroid hormone secretion or synthesis
- Defect in thyroid hormone transport
- Defects in thyroid hormone metabolism
- Thyroid hormone resistance
- Central hypothryoidism
- Transient congenital hypothyroidism
Newborn screening pre-requisites:
- Direct benefit to the neonate from early dianosis
- This benefit is reasonable balanced against financial and other costs
- There is a reliable test, suitable for neonatal screening
- There is a satisfactory system in place to handle testing, counselling, treatment and follow-up of positive screens.
Criteria for an effective population-wide screening test (e.g. on all
newborns):
- Availability of a test that is a) cheap and b) reliable
- Condition is treatable, has high morbidity, and preferably is reasonably common
Criteria required before performing ante-natal diagnostic tests:
- A reliable and safe diagnostic test is available
- There is a significant risk of the foetus having the suspected IMD.
- Condition is serious enough to warrant offering termination and is not treatable.
- Parents are willing to consider termination of pregnancy if foetus is shown to be affected.
WHO screening criteria (2008)
- The screening programme should respond to a recognized need.
- The objectives of screening should be defined at the outset.
- There should be a defined target population.
- There should be scientific evidence of screening programme effectiveness.
- The program should integrate education, testing, clinical services and programme management.
- There should be quality assurance, with mechanisms to minimize potential risks of screening.
- The program should ensure informed choice, confidentiality and respect for autonomy.
- The program should promote equity and access to screening for the entire target population.
- Program evaluation should be planned from the outset.
- The overall benefits of screening should outweigh the harm.
Newborn screening strategies:
- Test TSH with a reflex T4 if TSH is high
- T4 with reflex TSH if T4 low
- Heel prick (day 2-5 of life) vs cord blood
- Repeat screen at 2-6 weeks?
- Confirmation tests TSH + T4 or fT4
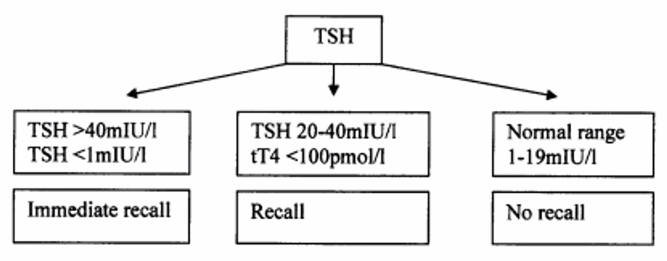
Problems with congenital hypothyroidism screening in South Africa
- Long recall success and high default rate
- Long delays before review and to starting treatment
Only 34% patients are contactable in SA after testing positive (audit by Dr. Michelle Carrihil). Many patients who were recalled – still defaulted.
Treatment
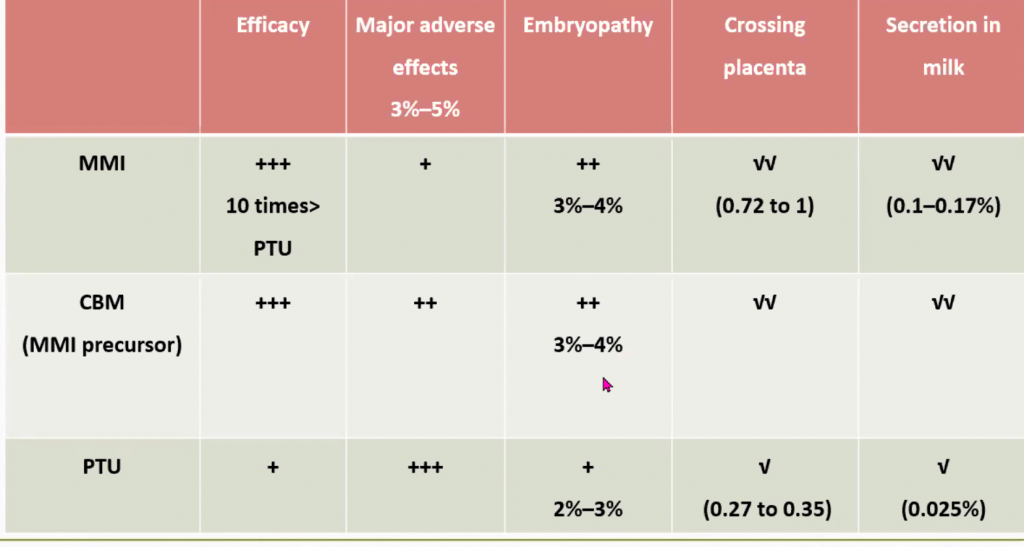
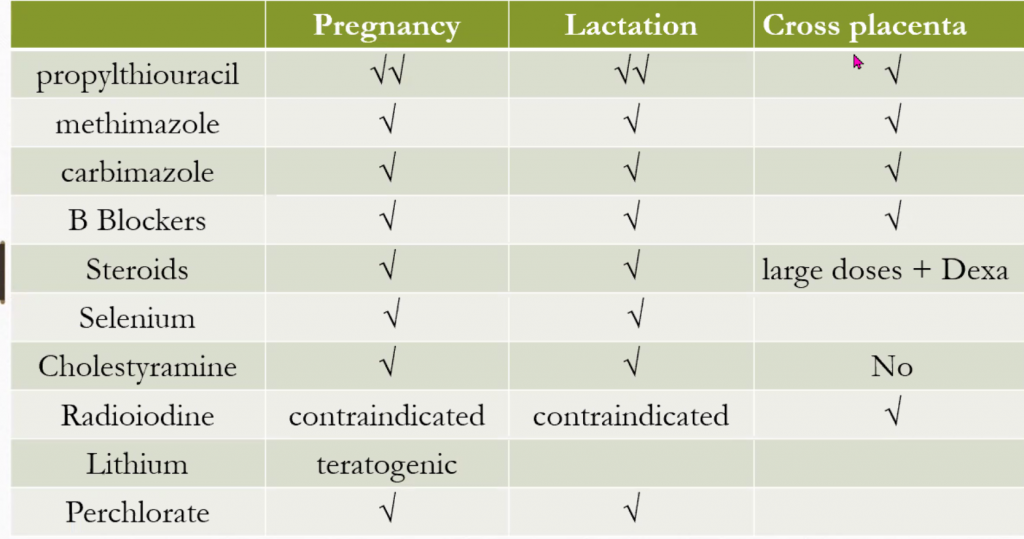
Levothyroxine is the Rx of choice. The aim is to raise T4 and normalise the serum TSH. If given withing the first 2 weeks of life, it can prevent intellectual impairment in >90% of cases.
Conclusion
- This patients intellectual impairment and the associated social implications of this could have been prevented with an effective screening programme.
- Screening programmes despite being projected to be effective, can have many unforeseen difficulties
- Auditing screening programmes regularly and implementing improvements is a necessary part in ensuring its efficacy
References
- Carrihill, M. 2008. An audit of the thyroid screening programme in the Peninsula Maternal and Neonatal Services. University of Cape Town.
- https://www.who.int/bulletin/volumes/86/4/07-050112/en/
- Uptodate section on congenital hypothyroidism
- BURTIS, C. A., ASHWOOD, E. R., BORDER, B., & TlETZ, N. W. (2001). Tietz fundamentals of clinical chemistry. Philadelphia, WB. Saunders.
- Medscape section on congenital hypothyroidism