A case of crystals in the cornea, but wait first…
| HOSP # | WARD | Red Cross Padiatric Hospital | |
| CONSULTANT | George van der Watt / Surita Meldau | DOB/AGE | 1y 5m |
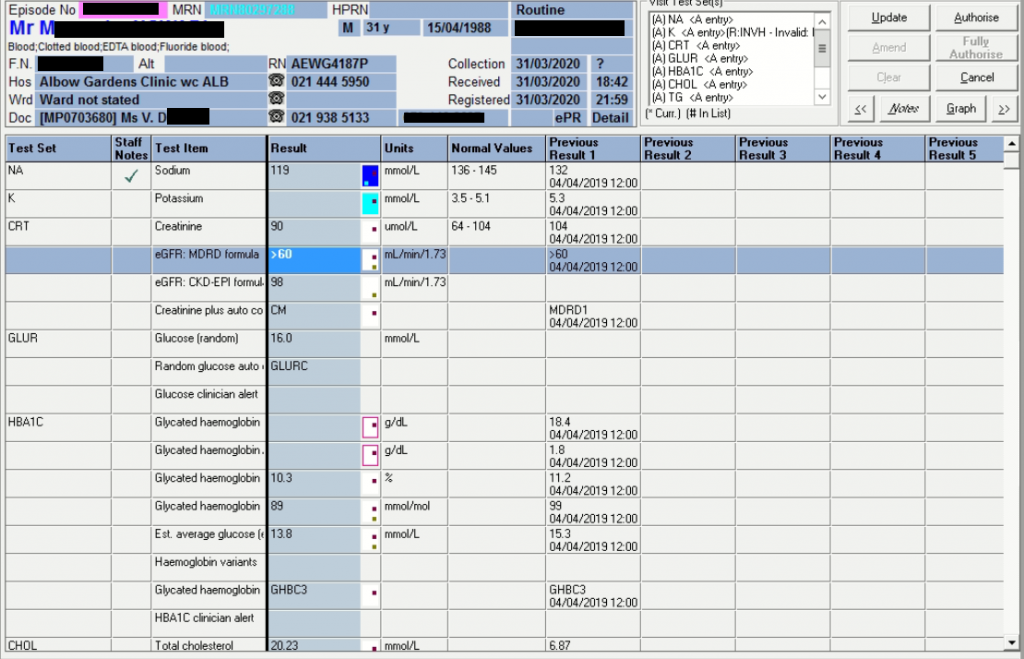
Abnormal Result
Inorganic phosphate 0.85 L mmol/L (1.00 – 1.95)
Presenting Complaint
It is unknown what this patient’s presenting complaint was.
Common complaints in similar patients are:
- Photophobia (see slit lamp picture below, which explains why).
- Bone pain
- Weakness

History
The items below illustrates the recorded information on the laboratory request forms as they were captured by our lab staff on the respective samples:
SEPSIS
Vit D Deficiency
Vit D Deficiency
HYPOCALCAEMIA
?LOSSES
?PTB
?PTB
?FANCONI
?SEPSIS
?SEPSIS
?SEPSIS
? FANCONI SYNDROME
? Cystinosis
Risk factor:
Contact
Cough
Fanconi's ? cystinosis
Hypocalcaemia
Pneumonia.
Pneumonia.
Fanconi syndrome
? Hypoglycaemia
post iv calcium for hypo
Sepsis. Cystinosis
Sepsis. Cystinosis
Cystinosis
FANCONIS SYNDROME
Fanconi's syndrome
Cystinosis
Cystinosis
Sepsis. Fanconi syndrome, hypocalcaemia
Hypocalceamia
CYSTINOSIS- HUNGRY BONE DIt can be seen above that the clinicians were noting Fanconi’s syndrome and were querying Cystinosis.
Examination
Data not available. Generally patients with Fanconi’s syndrome present with loss of electrolytes from the proximal and distal renal tubuli:
- Nephropathic cystinosis in untreated children is characterized by renal Fanconi syndrome, poor growth, hypophosphatemic/calcipenic rickets, impaired glomerular function resulting in complete glomerular failure, and accumulation of cystine in almost all cells, leading to cellular dysfunction with tissue and organ impairment. The typical untreated child has short stature, rickets, and photophobia. Failure to thrive is generally noticed after approximately age six months; signs of renal tubular Fanconi syndrome (polyuria, polydipsia, dehydration, and acidosis) appear as early as age six months; corneal crystals can be present before age one year and are always present after age 16 months. Prior to the use of renal transplantation and cystine-depleting therapy, the life span in nephropathic cystinosis was no longer than ten years. With these interventions, affected individuals can survive at least into the mid-forties or fifties with satisfactory quality of life.
- Intermediate cystinosis is characterized by all the typical manifestations of nephropathic cystinosis, but onset is at a later age. Renal glomerular failure occurs in all untreated affected individuals, usually between ages 15 and 25 years.
- The non-nephropathic (ocular) form of cystinosis is characterized clinically only by photophobia resulting from corneal cystine crystal accumulation.
Because the thyroid glands are actively translating and resorbing thyroglobulin, the thyrocytes (thyroid colloid epithelial cells) are prone to accumulation of cysteine within their lysosomes, hence these children often also develop hypothyroidism with a palpable thyroid gland.
Laboratory Investigations
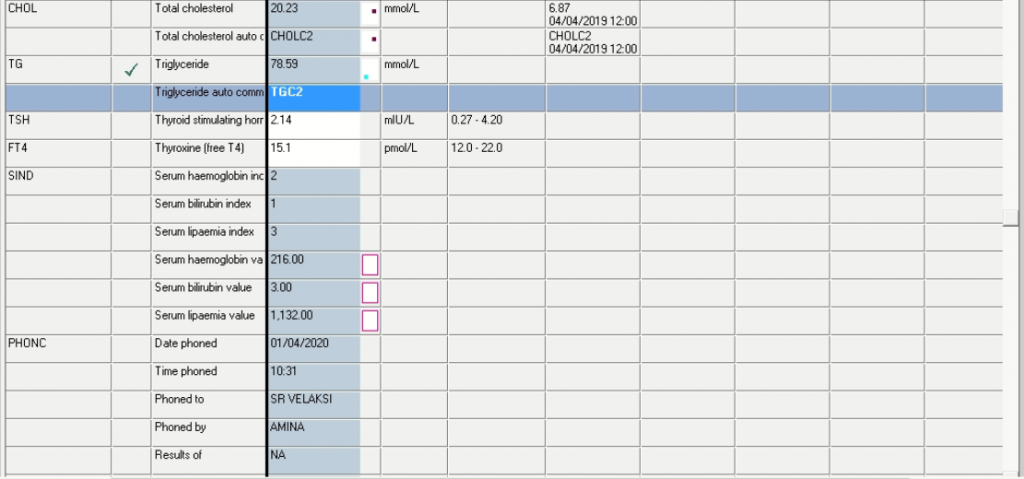
Biochemistry:

Newest results are to the left, oldest on the right.


Genetic screening
PCR with enzyme digest:

The PCR product before restriction enzyme digest is 261 bp long.
Without the mutation, the restriction enzyme will cut this product in 2 places, yielding 3 fragments 179, 42 and 40 bp. (Normal)
With the mutation, the restriction enzyme will cut the PCR product in 3 places, yielding 4 fragments, 135, 44, 42 and 40bp.
1st PCR with enzyme digest:

1. Patient sample
2. Positive control
4. Normal (Negative) control
5. Blank
Upon the first PCR for a gene screen which was done, there occurred some evaporation in the third tube (lane no. 3 – positive control), upon incubation with the enzyme digest mix. This incubation step is minimum 3 hours at 37 degrees Celcius and do not have the high temperatures associated with the PCR process. Thus the reason for evaporation remains unclear and may have been an incompletely closed lid or a defective PCR tube rim or cap.
It is not expected to see an undigested PCR product (>200bp) as can be seen in the top band in lane 3, hence it is clear that there was incomplete digestion of PCR product in this PCR tube. Even though it is clear that the patient sample ( lane 1) represents a homozygous positive result (lane 2), one cannot authorize results when both the positive and normal control worked well.
Thus the PCR was repeated.
2nd PCR with enzyme digest:

1. Patient sample
2. Positive control
3. Positive control with 2x enzyme mix added
4. Normal control
5. Blank
Image intentionally left uncropped and long vertically: Note the cloudy grey portion below the bottom most bands **see “Take Home Messages” below
In the second PCR it can be well seen how the Positive control did undergo complete digestion. No evaporation was noted in this run.
Other Investigations
Leucocyte cystine is another investigation which helps make the diagnosis in patients with presumed cystinosis.
Leucocyte cystine report in this patient:
Protein 0.58 g/L
Leucocyte Cystine 1.16 nmol/mg protein
Reference range:
- Normal < 0.1 nmol cystine/mg protein
- Cystinosis > 1.0 nmol cystine/mg protein
- Cystinosis on Rx (target level) < 0.5 nmol cystine/mg protein
Our reporting comment on the Lab Information Systems reads as follows:
Please note that the diagnosis of cystinosis can be confirmed in the majority of South African patients by screening for the common South African Black mutation CTNS-c.971-12G>A which results in an estimated newborn incidence of 1/10 000 in this population. A molecular diagnosis is of value in that siblings of index cases can be screened and identified for early intervention which improves the outcome in this disorder.
Final Diagnosis
Patient is homozygous positive for cystinosis by the common South African mutation, as confirmed on leucocyte cystine as well as on the gene screen.
Take Home Message
** The grey portion at the bottom of the electropherogram – this indicates the movement of the ethidium bromide out of the gel towards the cathode. One should ideally not let a gel run beyond the intended time period as the migration of ethidium bromide “dye front” beyond the smallest band may cause band to “de-stain” and not be visualized well.
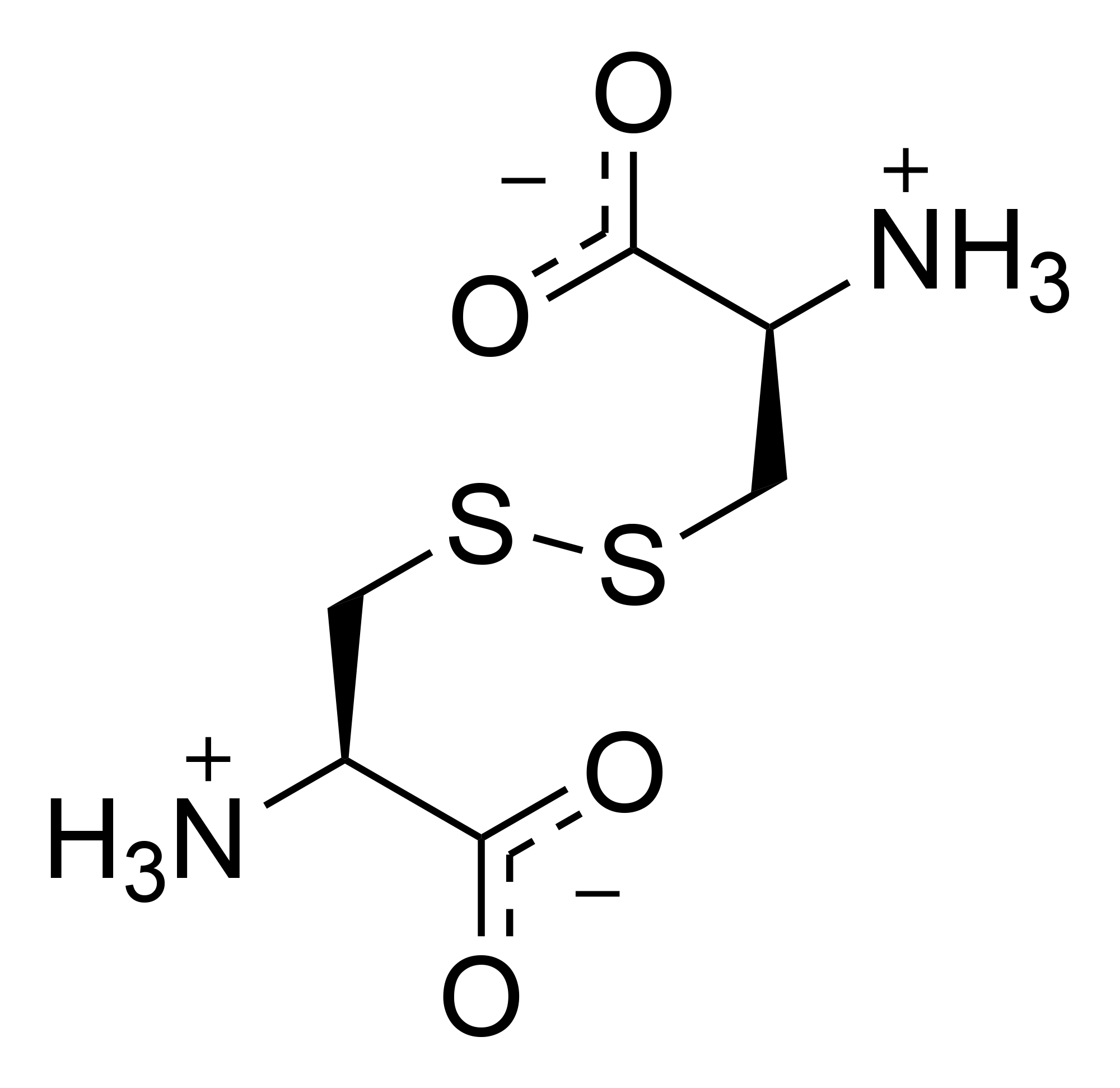
The CTNS gene provides instructions for making a protein called cystinosin. This protein is located in the membrane of lysosomes. Proteins digested inside lysosomes are broken down to amino acids. These are then moved out of lysosomes by transport proteins. Cystinosin is a transport protein that specifically moves the amino acid in its dimeric form cystine out of the lysosome.
More than 80 different mutations that are responsible for causing cystinosis have been identified in the CTNS gene. The most common mutation is a deletion of a large part of the CTNS gene (sometimes referred to as the 57-kb deletion), resulting in the complete loss of cystinosin. This deletion is responsible for approximately 50 percent of cystinosis cases in people of European descent. Other mutations result in the production of an abnormally short protein that cannot carry out its normal transport function. Mutations that change very small regions of the CTNS gene may allow the transporter protein to retain some of its usual activity, resulting in a milder form of cystinosis.

The treatment / management of patients with cystinosis includes Cysteamine, a drug which binds cysteine and forms Cysteamine-cysteine (see figure above). This molecule is similar in structure to lysine and can be exported from the lysosomes by a lysine transporter.
Interestingly, South Africa likely has the highest incidence of cystinosis in the world due to a common mutation, G > A mutation in intron 11 of the CTNS gene (c.971-12G > A p.D324AfsX44), likely due to some sort of founder-effect in black and coloured patients: https://link.springer.com/article/10.1007/s00467-014-2980-7 I’m proud of this article as it was published by scientists at our institution.
A useful web site to learn nomenclature of gene variants is HGVS.